What's New
Search
Conferences
Public Domain
SDPD-Database
VRML
STRUVIR
ARIT
ARITVE
RMCAW95
Links
Eccentric
Also
Hits
Overview
Mirrors
CV
Citations ISI
ARRED
Colorado Springs, Colorado, USA
August 3-7, 1998
New Developments in Microstructure Analysis via Rietveld Refinement
A. Le Bail
T1 - Innovative applications of the Rietveld method are likely to include new developments in microstructure analysis. When the minimal full width at half maximum is as low as 0.01° (2q ) as with some synchrotron data, sample effects are no longer occulted by instrumental effects. Finer microstructure details are revealed, needing adapted Rietveld software. Recent innovations and applications are discussed in this conference, with a special emphasize on anisotropic effects in the Rietveld method.
{kind=link}
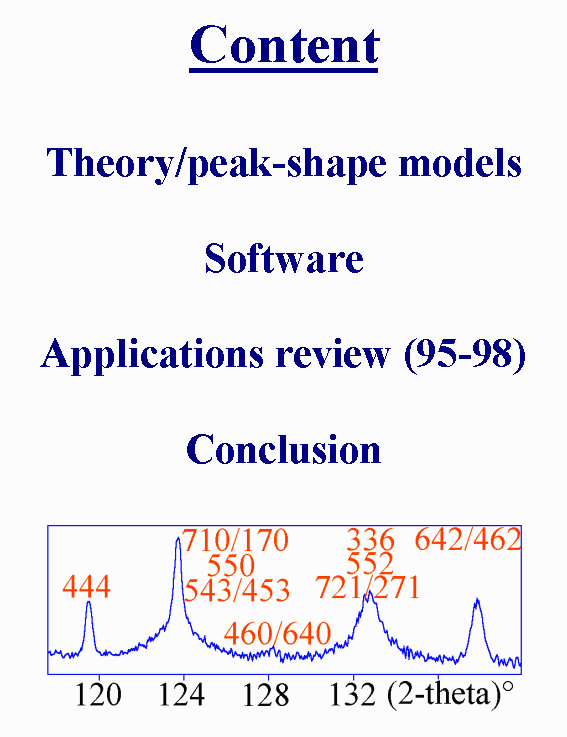
T2 - We will shortly have a look at the theory and peak-shape models, then software will be listed and recent applications will be reviewed.
{kind=link}
T3 - The experimental profile h is the convolution of contributions from the wavelength distribution, the instrumental and the sample profiles, respectively g and f. The g profile is modelled in the Rietveld method according to three different approaches. In the so-called "Fundamental Parameters Approach", the various physical phenomena responsible for the instrumental and wavelength contributions are each of them modelled by mathematical functions, for instance by sums of Lorentzians, and the final calculated profile is built by their numerical convolution. In the most generally used approach, profile functions are chosen in order to best fit the data, by using more or less simple analytical functions : pseudo-Voigt, Pearson VII, possibly splitted (...), this may be called the analytical profile approach. The third way for profile shape description consists in the learnt-profiles approach, applied in programs like XRS, ARIT1, in which profiles are parametrized (for instance by Fourier coefficients) by using experimentally observed ones.
{kind=link}
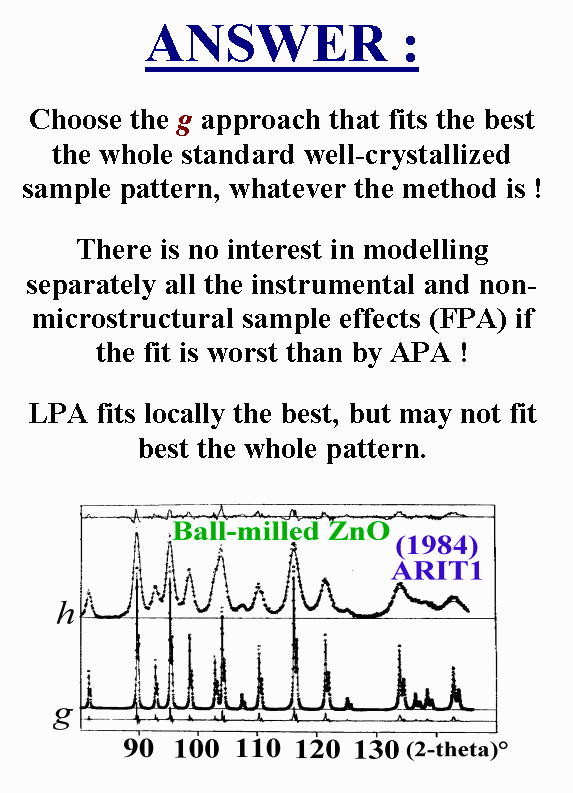
T4 - Which approach should be used ? If we consider the microstructure point of view, In the more exact version of the Warren-Averbach method, the profiles should never be modelled, but experimentally measured and then deconvoluted. No modelling at all is the best approach. In this sense, pretending that the Fundamental Parameter Approach is better than the Analytical Profile Approach is nonsense because none is as good as measuring profile shapes. Learning profile is almost equivalent to measuring. However, the problem of learnt-profiles is that they apply perfectly only at the angular position from where they are parametrized. Modelling comes back for their extrapolation to the whole powder pattern. In my opinion, no matter if the whole g profile is modeled by taking account of its various components independently or simultaneously respectively by FPA and APA. Both methods are in fact approximated approaches. My recommendation would be to use the method providing the best fit on the g profiles, whatever it is.
{kind=link}
T5 - About using the appropriate g profile, let me show you how much the profiles can depend on the sample preparation. These two patterns were made from the same compound with the same recording conditions. One was filled in a side-loaded holder so that the surface sample was plane but some preferred orientation remains. The upper pattern was from a sample dusted through a sieve so that the surface was rough. If I had to deal with the microstructure effect of this sample and I wanted to treat simultaneously the structure constraint, which g function should I use ? That is a problem.
{kind=link}
T6 - For describing diffraction by any kind of matter, the Debye equation is the more general, which has yet been applied for modelling nanocrystalline solids, or two-dimensional materials, including turbostratically stacked layer systems. Nevertheless, the Debye equation cannot be directly introduced in the Rietveld method, that absolutely needs a hkl-based description of the diffraction phenomenon.
T7 - The Rietveld method is certainly not the best choice for a fundamental approach of microstructures, which turns to be the Warren-Averbach method. Extracting size and microstrain effects relative to a crystallographic direction by the W-A method needs first that the other broadening effects are removed by deconvolution. Then the size/microstrain effects can be extracted from a series of harmonic reflections. Unfortunately, the W-A method is applicable only if no overlapping occurs, in few trivial cases with high symmetry. Rietveld method users are now facing routinely mediumly complex structures with ten to fifty or more independent atoms in the asymmetric unit, for which not even a pair of harmonics is available without overlapping. This leads inevitably to model-based peak-shapes and semi-empirical approaches of which the double-Voigt approach is the more elaborated.
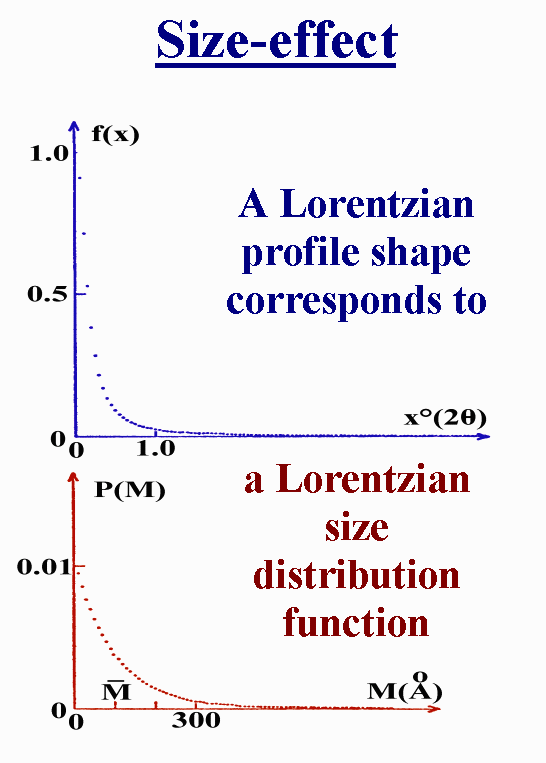
T8 - Are Voigt functions able to model any size effect ? That's OK for a Lorentzian profile shape which corresponds to a Lorentzian size distribution function. Note that such a size distribution presents the largest proportion of crystallites with the smallest size. I am not sure that it will represent well a general case, if any.
{kind=link}
T9 - But what about a Gaussian size distribution function like this one ? The corresponding profile shape has a shoulder and can be hardly modelled by a Voigt function. So that you see the point : even the Voigt function is not enough flexible. The same demonstration could be made for microstrain effect. Anyway, other means than those bad profile shape approximations have not been yet proposed that would apply more generally and could be included inside the Rietveld method.
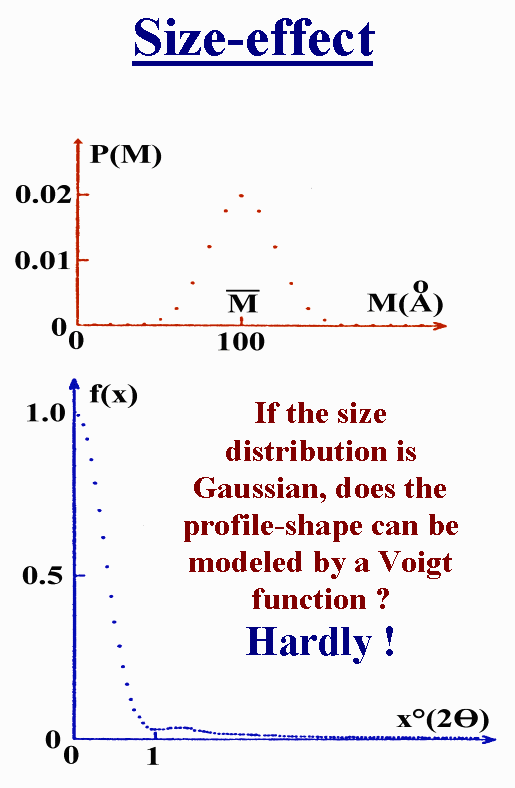{kind=link}
T10 - Here is a list of Rietveld softwares continuing to model size and microstrain effects by more or less empirical profile shapes. Even if some of them model the instrumental g profile by the so-called Fundamental Parameters Approach, none of them model the true-sample f profile by an equivalent fundamental approach. Nevertheless, implementation of microstructure approaches in Rietveld programs is improving continuously.
{kind=link}
T11 - As long as microstructure effects are isotropic, they can be accounted for easily in Rietveld refinements. Recent examples are the particle size characterization of magnesium and titanium oxides prepared by the sol-gel technique with different hydrolysis, by using DBW and WYRIET. The Thompsom-Cox-Hasting function allows the consideration of isotropic Lorentzian effect. Most of the times, anisotropic broadening is observed but not really treated.
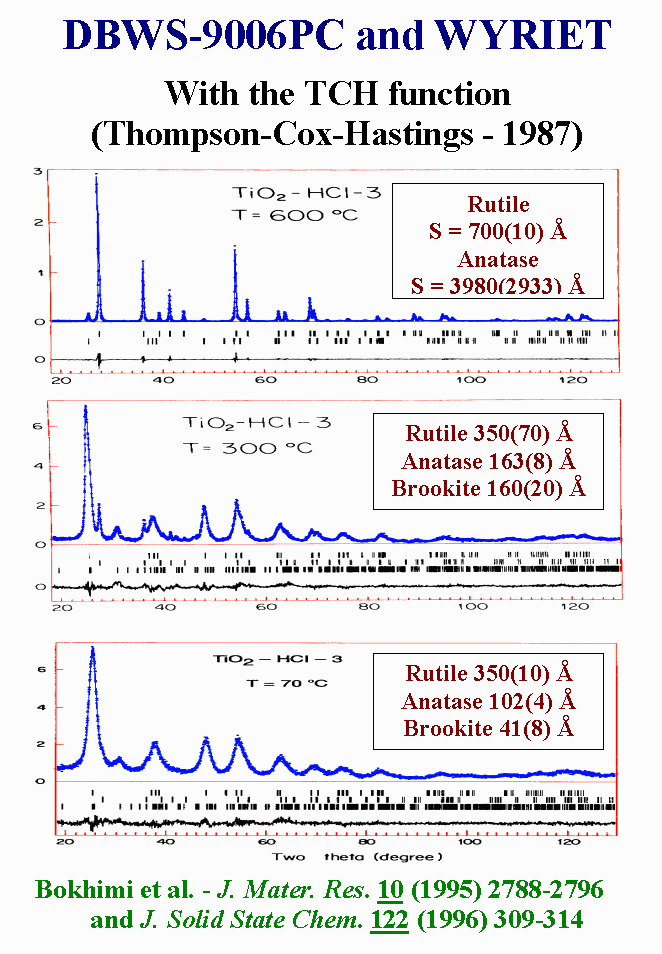{kind=link}
T12 - For instance, the Rietveld refinement of nanostructured hollandite powders could not converged well, due to such anisotropic effects associated with a fiber axis in the b direction. An improved fit was obtained from a powder pattern realized with a highly packed sample, inducing preferred orientation, and reducing the contribution of the narrowest reflections. A mean size of one-hundred and eight angstroms was proposed for the thickness of these needles. Dislocations were disclosed, but, not a word about microstrain can be found in this recent paper.
{kind=link}
T13 - More amazing is the ab initio structure determination of two polymorphs of cyclopentadienylrubidium in a single synchrotron powder pattern. Failing to index the pattern with a single lattice led to inspection of the peak width, that essentially fell into two populations, with Full Width at Half Maximum of 0.015° and 0.07°2q . With the hypothesis of the presence of two polymorphs, the two data sets were indexed and the structures were solved by direct methods applied to structure factors extracted by the Le Bail technique with FULLPROF. The program package GSAS was used for the final Rietveld refinements, but the paper did not concluded about microstructure of polymorph II, although the peak profiles were modelled by the Thompson-Cox-Hastings function. It was very probably an isotropic size or microstrain effect.
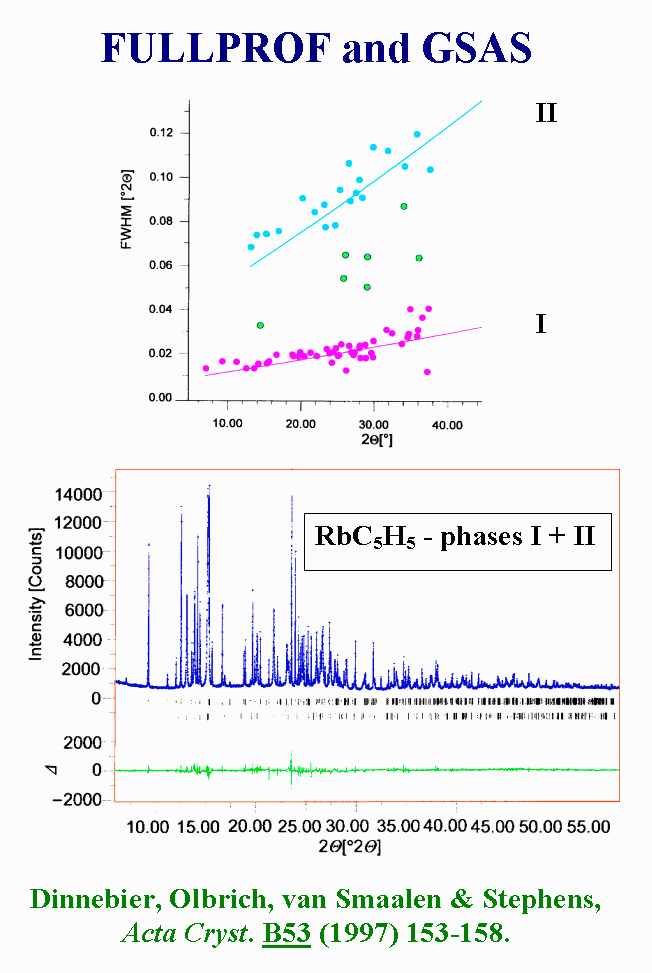{kind=link}
T14 - The ability of powder diffraction to reveal microstructures has been frequently questioned. A very interesting recent study of a Tantalum-Rhenium-Selenium material, that is an excellent lubricant, is exemplary. The Rietveld analysis with the help of the program RIETAN led to a superb fit. High Resolution Electron Microscopy image analysis revealed the coexistence of rotation twins, a high density of two-dimensional-defects in the (001) plane and prismatic cluster defects. So what ? The high density of two-dimensional defect seen by Electronic Microscopy is not enough for being seen by powder diffraction, possibly affected by a weak isotropic broadening, accounted for by a classical Rietveld refinement. In fact, the paper was not clearly explicit about a possible annealing difference between samples studied by microscopy and X-ray diffraction
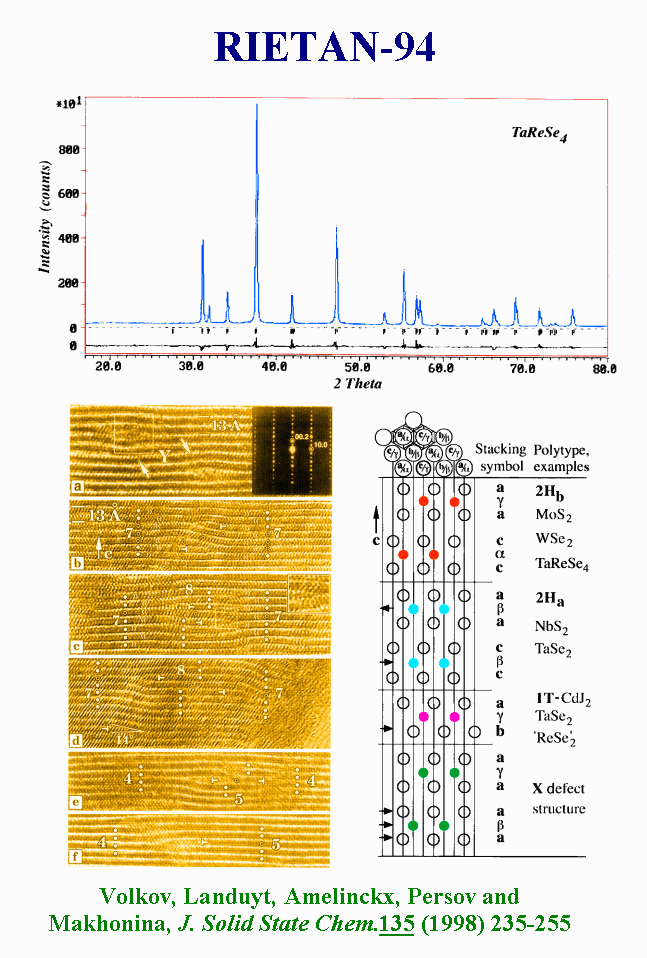{kind=link}
T15 - The Rietveld method was adapted (in DBWS) for the determination of the degree of crystallinity in semicrystalline materials. The crystalline peak profiles were modelled isotropically by pseudo-Voigt for a sample of polyethylene terephtalate. Background contributions were simultaneously optimized. In this study, the amorphous part comes from a measurement of a fully amorphous material. A different approach is possible for amorphous compounds that were modelled recently by the Rietveld technique, using a mean model, highly microstrained. An example is the quantitative analysis of silicate glass in ceramic materials that was realized by the Rietveld method (RIETQUAN), reusing the cristobalite-like model produced for silica glass by the ARITVE program.
{kind=link}
T16 - ARITVE is a multi-wavelength program allowing simultaneous refinement of neutron and X-ray data) devoted essentially to the simulation of diffraction patterns of amorphous or nanocrystalline compounds, by applying an extreme broadening to the reflections associated with a mean structural model. The most recent applications were to silica and some fluoride glasses. A new application to amorphous zinc-chloride is given here. All the available crystal structures of the glass-formers tetrahedral polymorphs were tested in order to fit the neutron pattern. The best fit was obtained from the d -zinc-chloride structure in which the glassy phase devitrifies. The refined parameters were only eight coordinates from fourteen thousands of reflections. The quite small resulting mean model was then used for the building of a larger box containing almost two-thousand atoms for a reverse Monte Carlo modelling. The original connectivity was constrained, so that no bond could be broken and no tetrahedra could be rearranged elsewhere. In this way, the Reverse Monte Carlo process could only slightly randomize the Zinc and Chlorine atom positions around their initial values. A good agreement was quickly obtained, very similar to this one. This result tends to show that the Rietveld for Disordered Materials and Reverse Monte Carlo methods can be reconciled with basically the same model. The model is mean and small by the Rietveld approach, it is large and statistically disordered by the Reverse Monte Carlo approach.
{kind=link}
T17 - We were up to now in isotropic microstrain and size approximations. Let us now examine a case with anisotropic line broadening, a lanthanum-nickel-hydrogen material, well and long studied by powder diffraction, including various Rietveld method programs. Each time, the conclusions about anisotropic microstructures were relatively poor, although taking them into account greatly improved the fit and the accuracy of the structural parameters. What is really behind the few values of direction-dependent size and microstrain values produced by these studies ?
{kind=link}
T18 - The defects induced by hydrogen absorption and desorption in the lanthanum-nickel compound were recently observed by Transmission-Electron-Microscopy, allowing to suggest what exactly is behind those mysterious root mean square strain and mean apparent size values estimated from powder data. The electron-microscopy study evidenced many kind of dislocations (ripple-like, fence-like, misfits, and loops), also anti-phase boundaries, fracture of slip band structure, structural disorder and micro-twins. All these defects have effects on the powder pattern that are gathered in those size and microstrain values. Progress and innovations in microstructure analysis by the Rietveld method will never overcome this fact : the powder pattern reveals the whole problem at a one-dimensional scale, and as a mean.
{kind=link}
T19 - The trend to deal qualitatively with microstructure effects is illustrated with the case of Norbornane. Fitting with isotropic line broadening was a kind of disaster. The synchrotron powder pattern was then fitted with the MPROF Rietveld program, using empirical simulation of anisotropic line broadening. In this approach, each usual U, V, and W parameters which describe the peak width and also the peak shape angular variation, was made hkl-dependent through second-rank tensors, leading to thirty-six variables maximum in the triclinic case. The RP value was lowered by almost a factor 2.
{kind=link}
T20 - Let us now examine some complex cases by using one of the most recent Rietveld programs. BGMN is a commercial package, that is able to behave as an expert system in a few predefined cases, including stacking faults in disordered layer silicates. It works by using a Fundamental Parameter Approach for the instrumental profile. About size-microstrain effects, BGMN uses Lorentzian broadening (for crystallite size) and squared Lorentzian broadening (for microstrain). BGMN has default dependence from size/strain. But you may select arbitrary other dependencies using the built in formula interpreter. This is what was done for kaolin by introducing a model of disordering : essentially the pattern is decomposed in 3 sub-phases with different broadening laws. Yes the fit is improved, but I have not found any size or microstrain or probability of stacking fault values in the published paper.
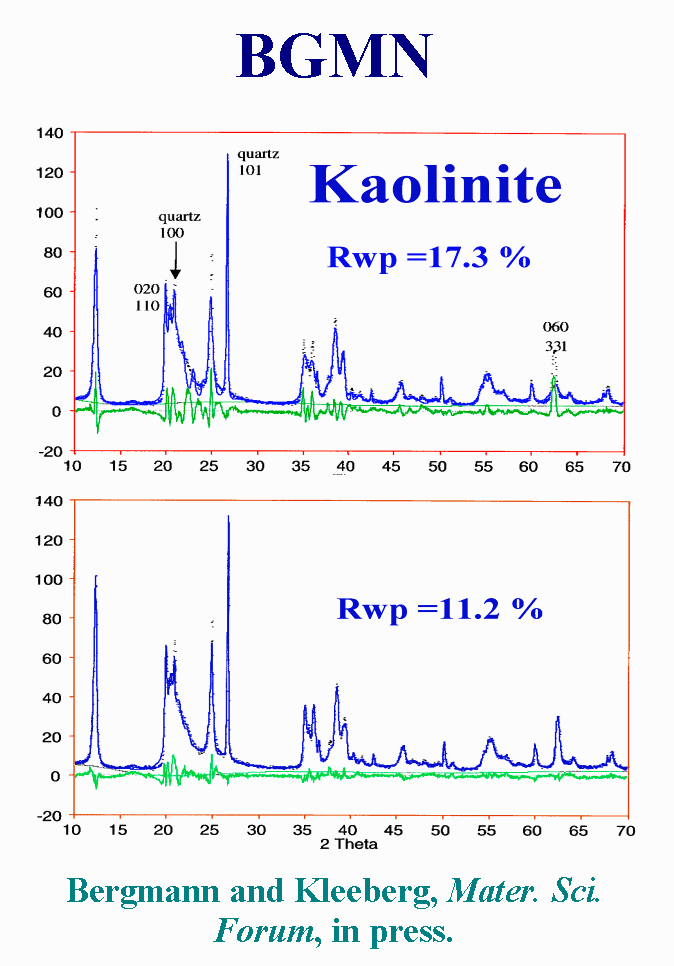{kind=link}
T21 - I asked Thomas Taut for some tests on my two favourite ill-crystallized samples with BGMN. One is lead oxalate of which the synchrotron pattern is hard to fit due to stacking faults. BGMN uses ellipsoids as a possibility for anisotropic effects, but this allowed to decrease the Rp value only by 2 %. Assuming that there were two slightly different real structures of the same phase, with nearly the same lattice constants but different peak broadening considerably improved the fit by BGMN with RP = 6.5 %. The approach is only qualitative but shows the ease of use of BGMN. In fact the fit was without the structure constraint, using the Le Bail method I guess, and could not be realized with the simultaneous refinement of the atomic coordinates.
{kind=link}
T22 - Another highly problematic powder pattern of a hydrogen niobium oxide was treated with BGMN in a similar way, using two subphases with different peak broadening, but with less success, the reliability RP value decreasing from thirty-five to eighteen percent, which is rather still high. Moreover, because the individual intensities are arbitrary distributed over the two "phases", introducing the structure constraint on intensities may alter considerably the fit quality. Both samples and patterns are challenges for the proposition of a physically sound model that would allow a satisfying fit by the Rietveld method, including the structure refinement. The perovskite-type niobium compound is very probably affected by anti-phase domains extending in the three dimensions as suggested by this picture.
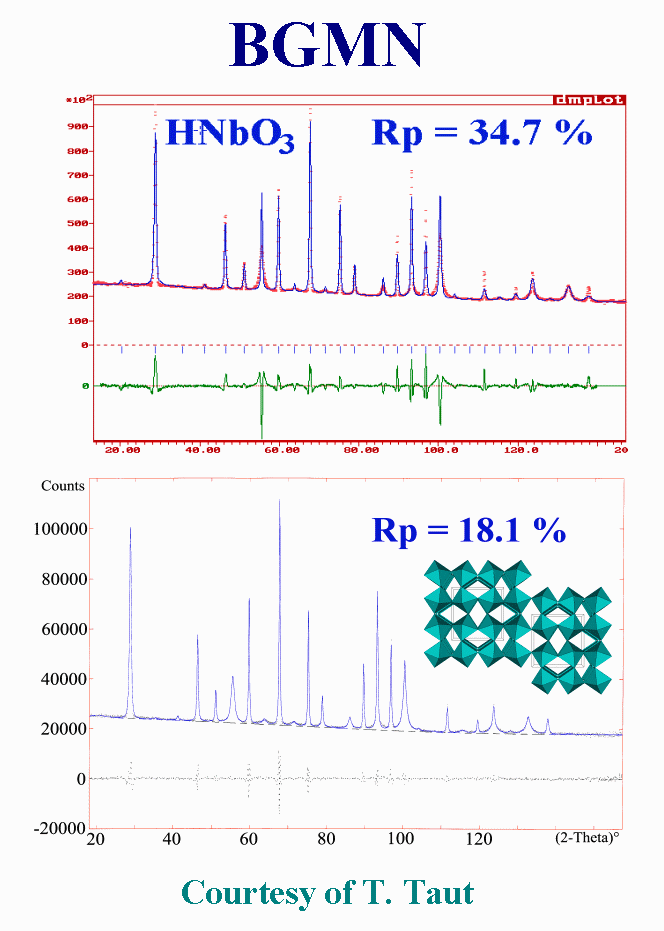{kind=link}
T23 - Most anisotropic approaches were done by using the ellipsoid description of size and microstrain. A recent phenomenological model of anisotropic strain broadening, proposed by Stephens, considers the distribution of lattice metric parameters within a sample. Each crystallite is regarded as having its own lattice parameters, with a multi-dimensional distribution throughout the powder sample. It is a generalized approach deriving from previous similar but more restrictive descriptions, already included in FULLPROF for instance. In the case of this sodium para-hydroxy benzoate, the reliability factor decreased from fifteen to eight percent.
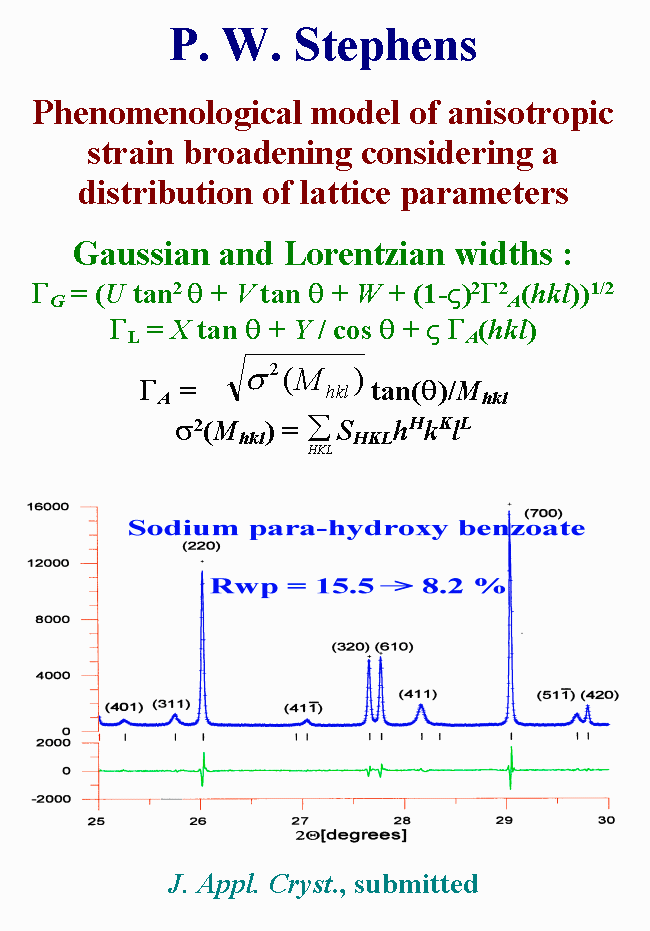{kind=link}
T24 - The Rietveld method broadens continuously its application domain, but, up to now, the microstructure approach lacks of flexibility and is mostly realized case by case. Expert systems have limited applications to few school cases like BGMN. Improving the accuracy of structure parameters is yet a sufficient target and justifies empirical approaches. Obtaining microstructure parameters having more recognized physical meaning is now desirable. For anisotropic effects, ellipsoids are progressively replaced by more general descriptions, in terms of spherical harmonics and distributions of cell parameters, allowing more complex cases to be accounted for. Unfortunately, the user's dream, which is to solve easily any problem through a friendly Graphical User Interface, is far from being realized. The problem (or possibly multiple problems) needs first to be identified.
{kind=link}
A preprint of the corresponding paper to be published in the conference CD-ROM (Advances in X-ray Analysis) is available on demand as a .doc file for MS Word 97, zipped with Winzip (denver.zip ~340Ko).
See also a more
recent conference on that topic.
Site in English OR French - Use Netscape 4.04 + CosmoPlayer 2.0 (VRML)
Copyright © 1995-1998 - Armel Le Bail