A. Le Bail, Laboratoire des Fluorures, Université du Maine, 72017 Le Mans Cedex, France.
New strategies using old formulae sometimes lead to efficient algorithms. Every user of the Rietveld method has worked with the so-called "Iobs" (or "|Fobs|") either by the simple presentation of the Bragg R factor or by Fourier difference calculation or so on. Some special uses of the Rietveld's formula for "|Fobs|" extraction were made (to be classified among the two stages alternatives to the Rietveld method), however the starting |F|s were always the |Fcalc|s corresponding to a (complete or partial) structure model (Toraya, Marumo and Yamase, Acta Cryst., 1984, B40, 145-150 ; Taylor, Miller and Bibby, Z. Kristallogr., 1986, 176, 183-192 ; Taylor, Z. Kristallogr., 1987, 181, 151-160).
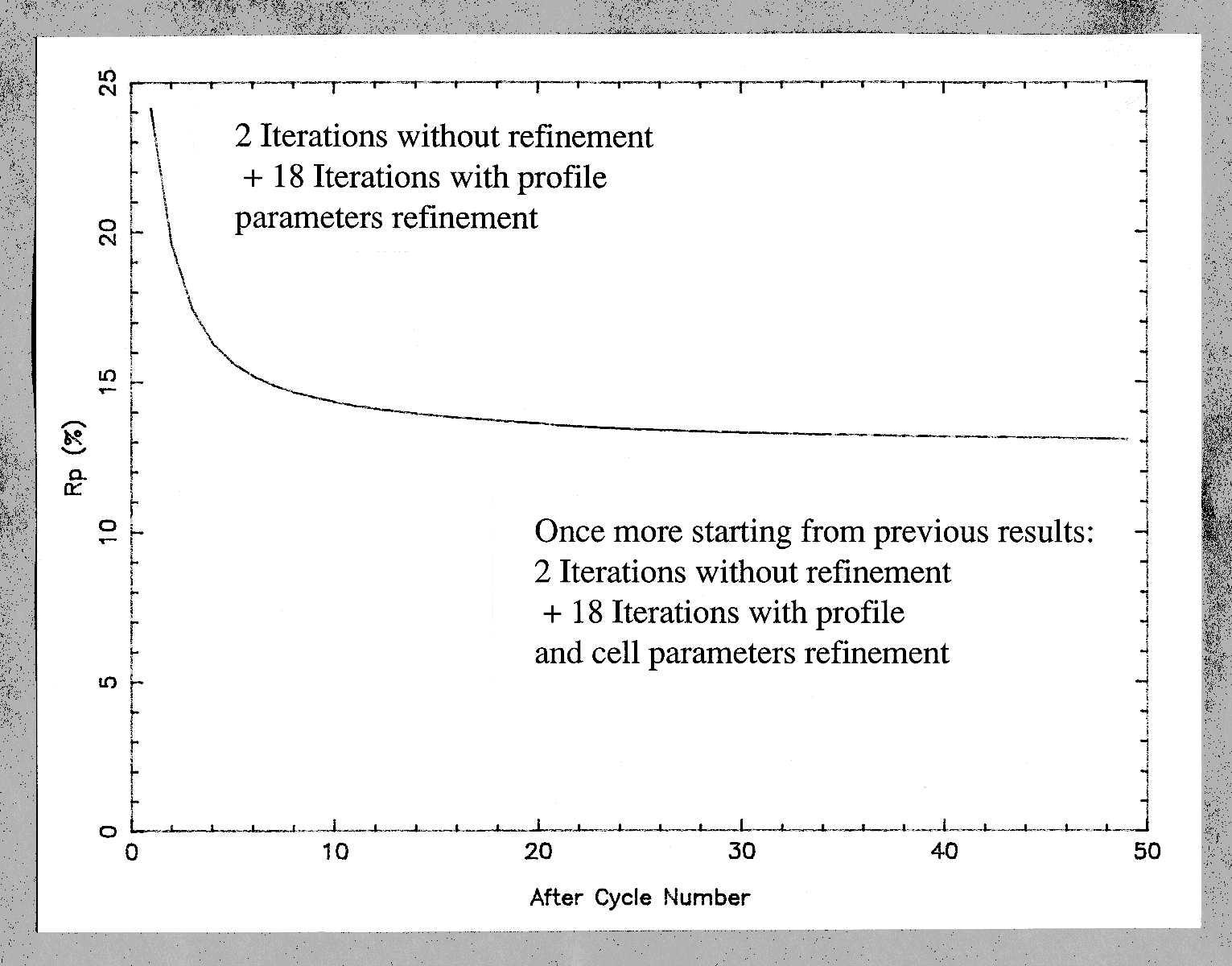
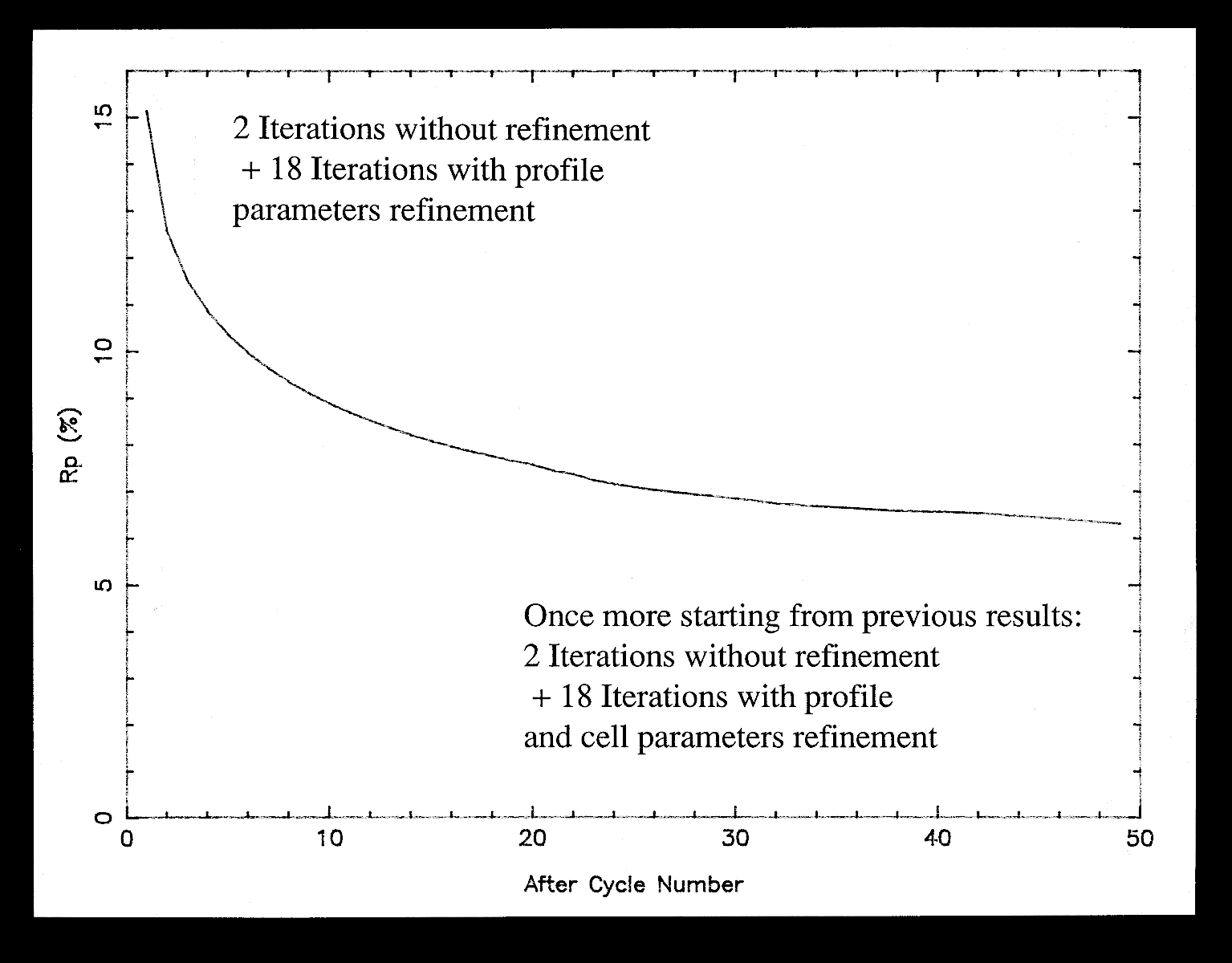
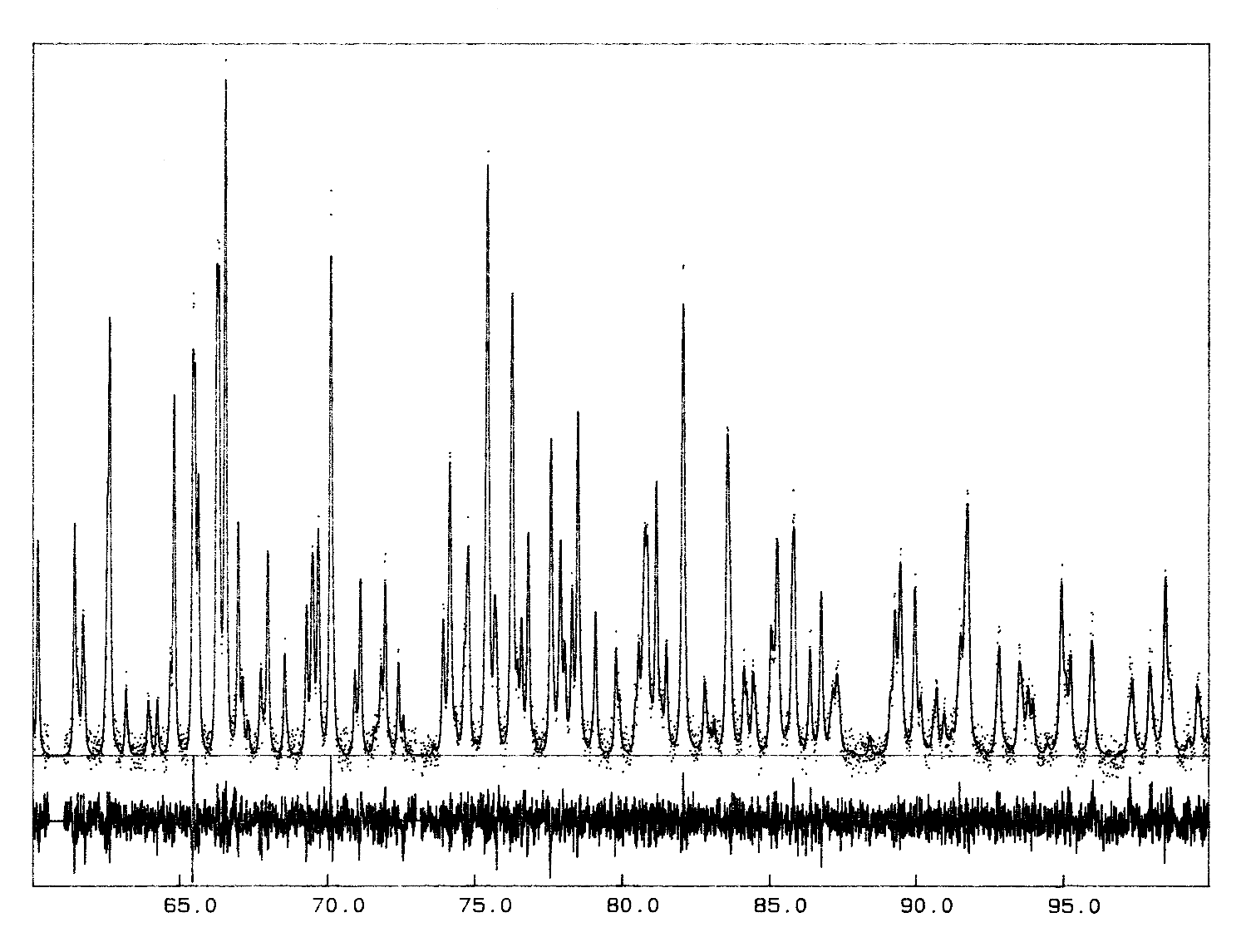
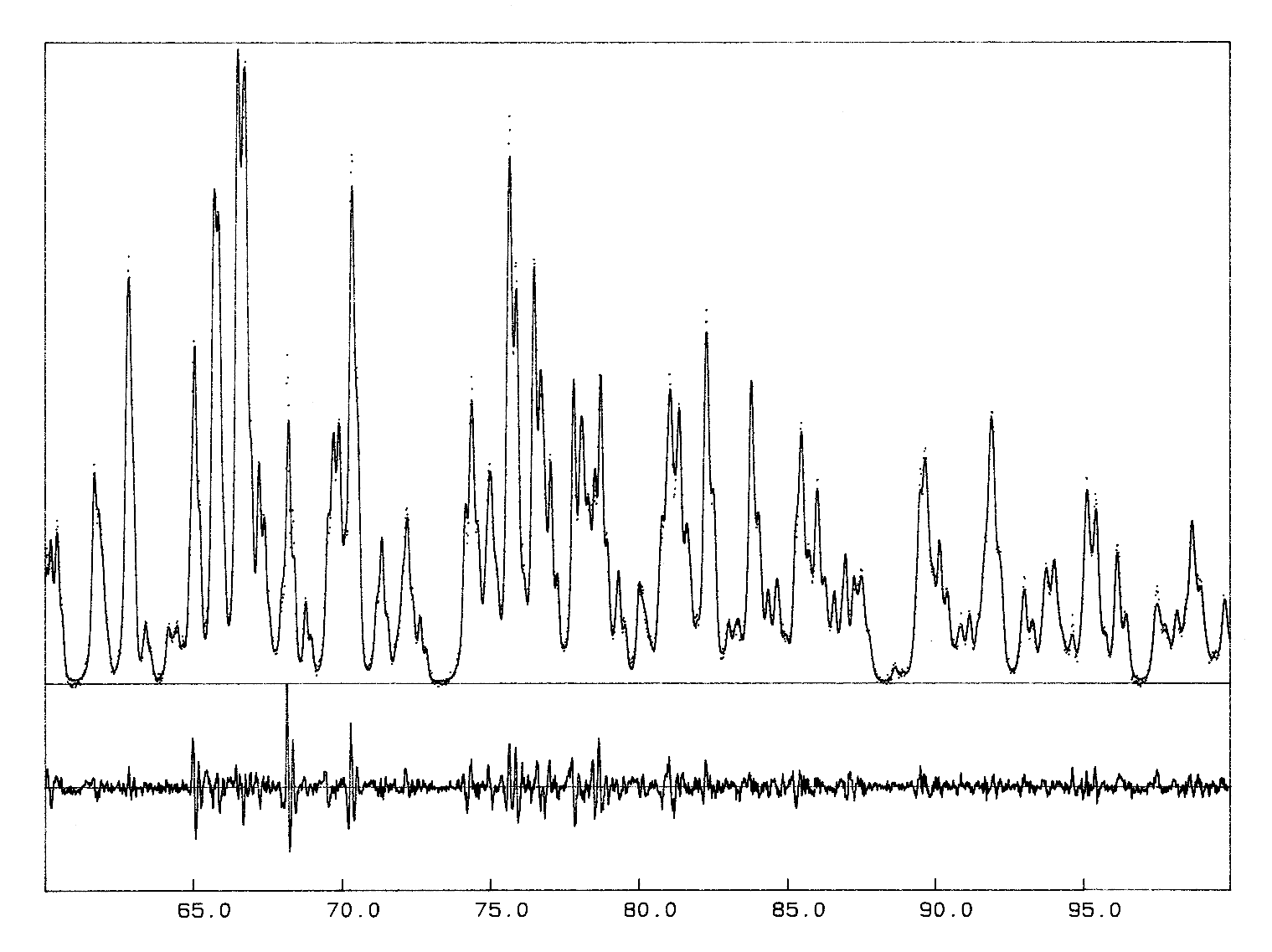
In the original Rietveld's work, the "|Fobs|" are estimated by partitioning among reflections contributing to each profile point of the pattern according to the |Fcalc|, thus they are biased. However, it will be shown that iterating the Rietveld's formula provides a nice and simple way to extract |Fobs| as accurately as possible, starting from initial |F|s arbitrarily set to have the same value. The first study applying such an algorithm for structure determination purpose (using the unpublished program ARITB) was from Le Bail, Duroy and Fourquet, Mater. Res. Bull., 1988, 23, 447-452. Near 20 ab initio structure determinations have been now performed in this way, either from conventional or synchrotron X-ray powder data. The algorithm has been implemented as an option in two other programs (FULLPROF, GSAS) ; any Rietveld program can be easily modified to accommodate this possibility to extract structure factors. The strategy necessary for success will be exposed. The best results are from cell and space group constrained conditions ; only all equal starting |F|s can ensure that the structure factors of strictly overlapping reflections are undifferentiated : they keep the same value after each iteration ; lt is strongly recommended to start from very precise cell parameters (not approximated ones) in order to avoid false minima in case of complicated patterns with strong overlapping ; some iterations are necessary without modifying cell and profile parameters in order to start their least-squares refinement at further cycles in good conditions. A definite advantage of the procedure is the very low number of parameters to be refined (15 maximum in ARITB) allowing it to handle problems of any size. Extracting in one run the several thousand |F|s (for instance from synchrotron powder data with 0.020° 2-theta-FWHM) implicit for large and/or low symmetrical cells is not utopia.
Accuracy in Powder Diffraction II
Proceedings of the International Conference, May 26-29, 1992
NIST Special Publication 846, E. Prince and J.K. Stalick, Editors
page 213.
|
|
|
|
|
|
|
|
||||
| a test case |
|
|||
|
|
Results |
|
|
|
|
|
EXTRA
|
CTING
versus iterative number |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
SOL
|
VING | ||
|
|
|
|
||
|
|
|
|||
|
|
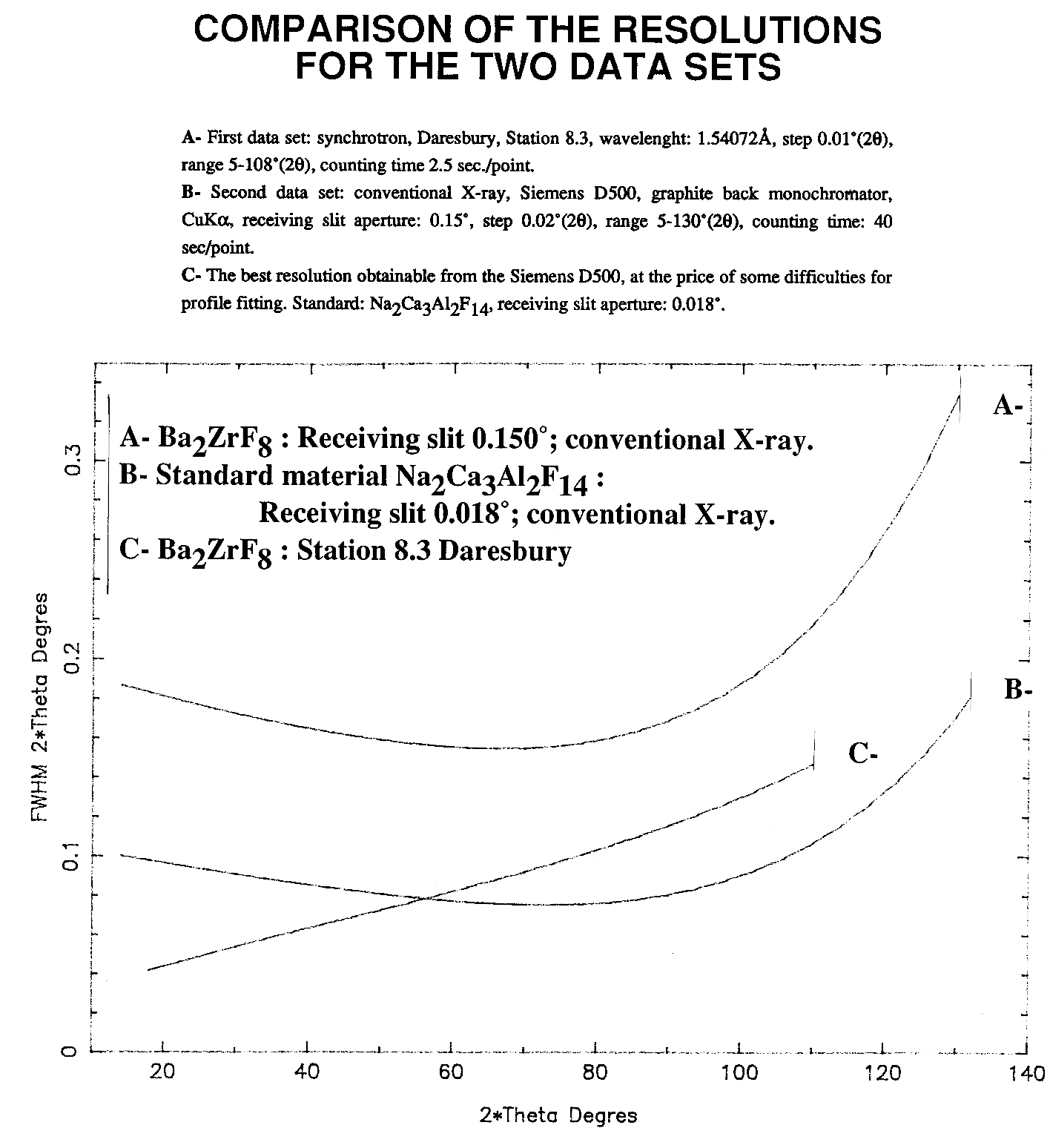
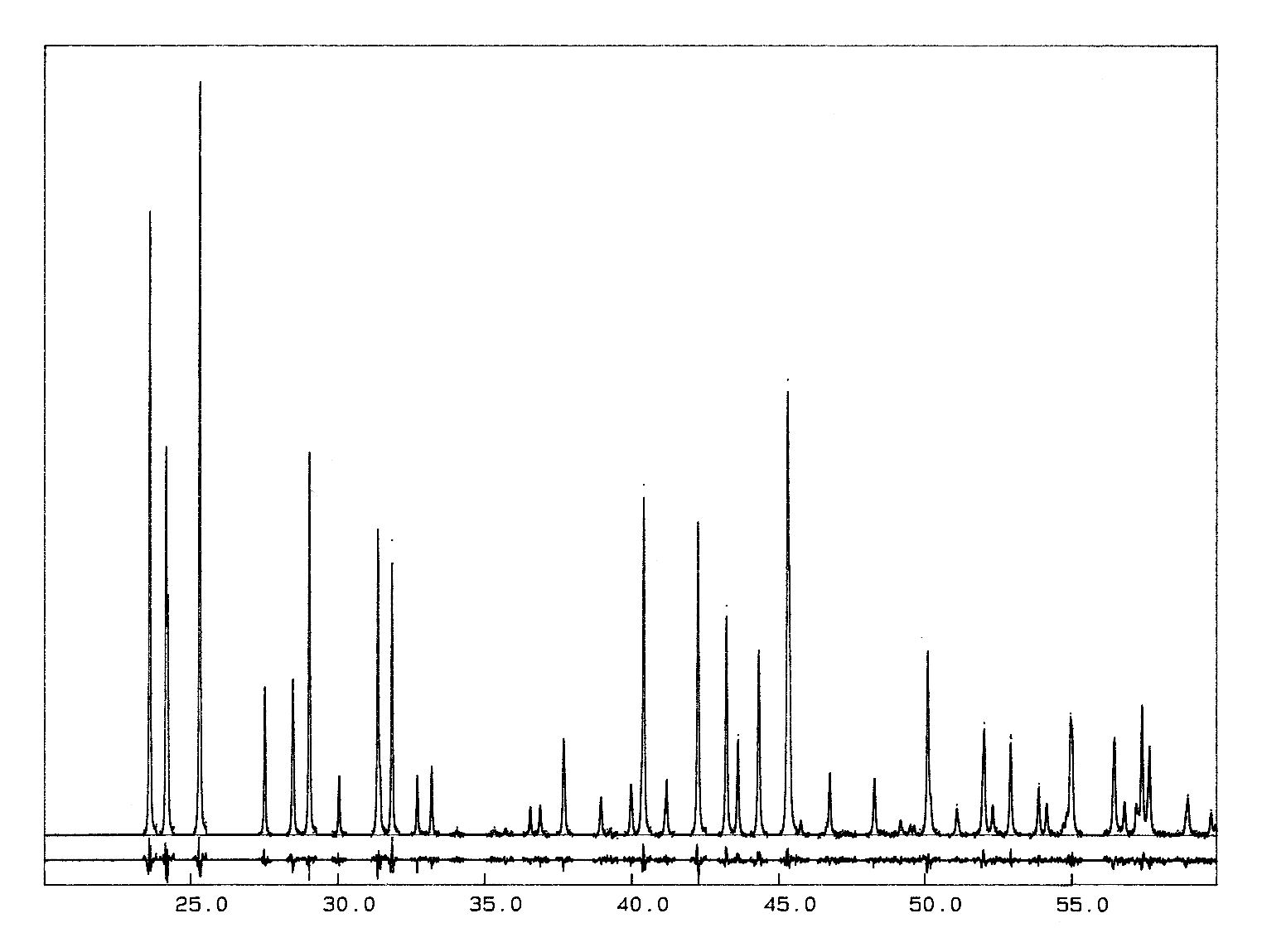
The crystal structure of Ba2ZrF8 described in
that powter as an example was published much later (1998 !), waiting for
some neutron data too, for having more weight and accuracy on the F atoms
:
A. Le Bail and J.-P. Laval, Synthesis and crystal structure of alpha-Ba2ZrF8
and Pb2ZrF8 determined ab initio from synchrotron
and neutron powder diffraction data, Eur. J. Solid State Inorg. Chem.
35 (1998) 357-372.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}