Full conference text and slides (see also the previous conference text):
T21 - The SDPD Round Robin was organized by Lachlan Cranswick from the CCP-14, Daresbury, and me, following some public discussions at the Rietveld mailing list about structure determination by powder diffractometry. This Round Robin had to be fast and was based exclusively on Internet means. Data and questionnaires were available from a Web site starting from May 18 and the deadline was the last day of June. It was copiously announced on many Newsgroups and Mailing lists related to crystallography, material science, structure prediction and so on. More than 800 visitors had a link to the Homepage, of which 70 downloaded the data.
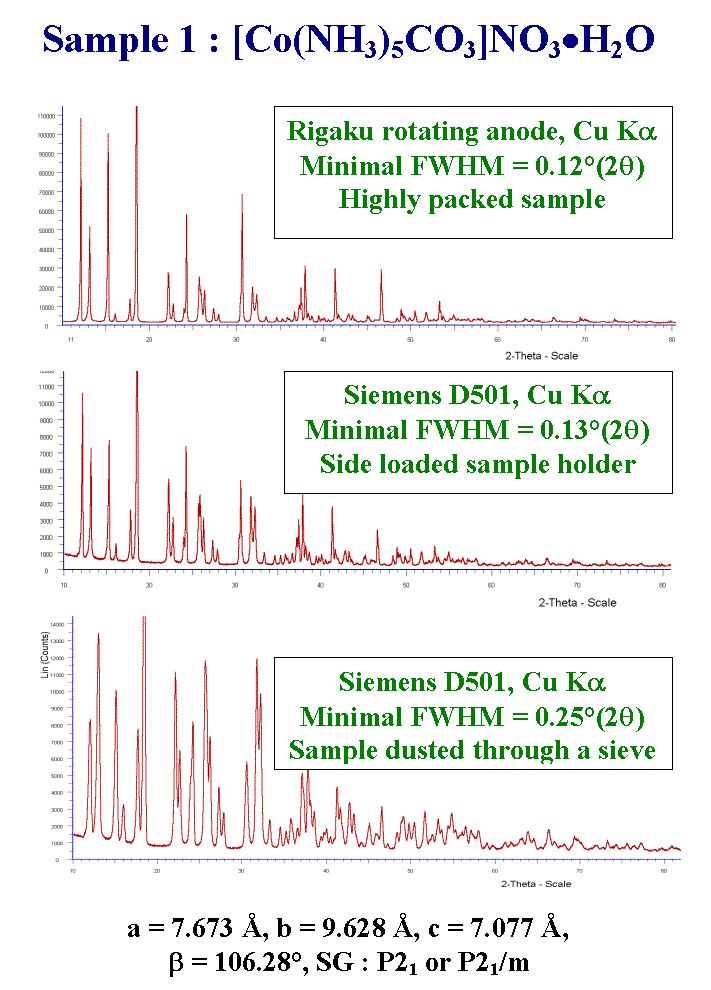
T22 - The powder patterns for 2 samples were provided. Here is the first one, inorganic. Cell parameters and possible space groups were provided together with 3 powder patterns showing preferred orientation at various degrees. It was expected that the developpers of methods taking advantage from preferred orientation would find interesting to participate. It was clear that we were focusing on the solution stage and not on the chemical analysis or indexing or even Rietveld refinement stages.
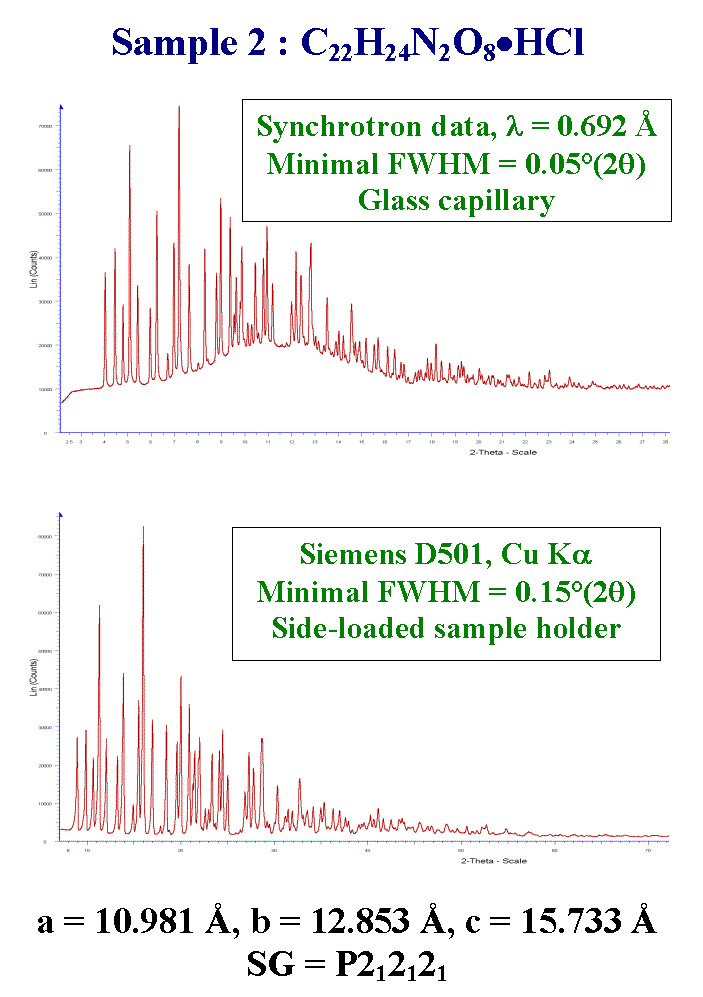
T23 - Here is the data for the pharmaceutical second sample. Cell parameters and space group were given. A nice synchrotron pattern was provided as well as a conventional X-ray powder pattern with similar resolution, due to the use of a longer wavelength. That's all we provided.
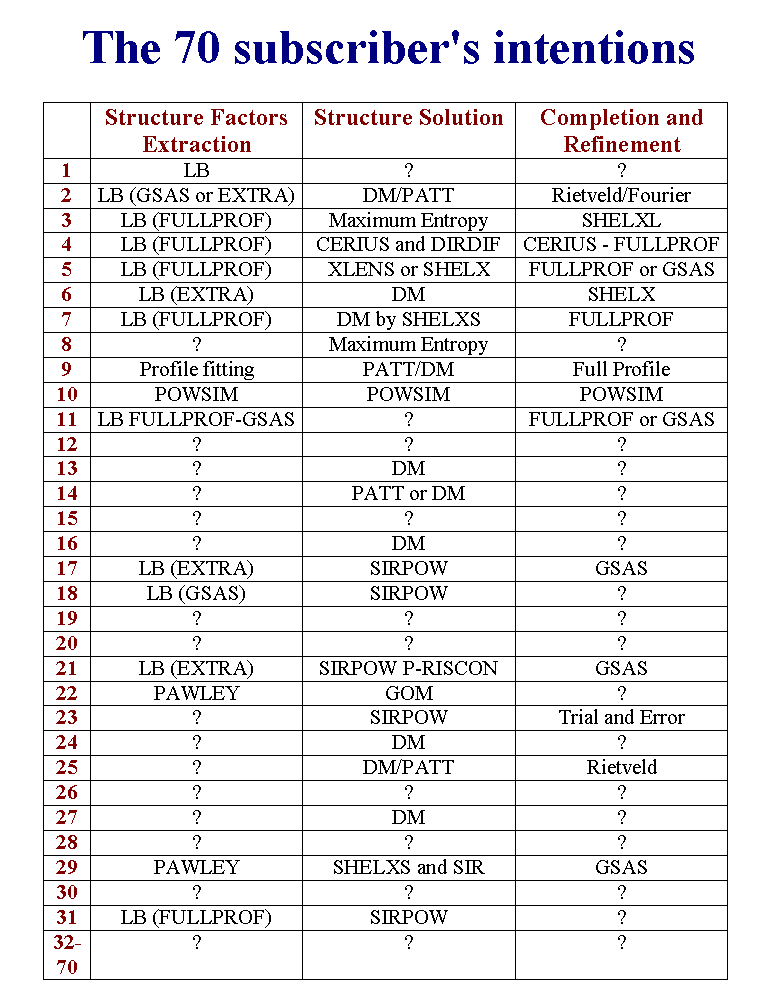
T24 - The 70 peoples having downloaded the data may be considered as subscribers of this Round Robin. In fact the possibility was given for either anonymous download or filling a Web form asking for details about which methods and software will be used for 3 steps : structure factors extraction, structure solution and structure completion and refinement. Only 31 subscribers filled in the Web form, more or less completely. One can recognize the most frequently used softwares : GSAS, FULLPROF, SHELX, SIRPOW and so on.
T25 - We received finally only 5 filled questionnaires from 4 final participants. One for sample 1 and 4 for sample 2. Participant 1 made a very fast reply but he was unable to provide coordinates. By a search in the Cambridge Structural Database, he found easily the reference for the pharmaceutical compound as being the tetracycline hydrochloride. Then he suggested that the coordinates should be found in this reference. Unfortunately, the coordinates are not available in this paper, nor in the Cambridge Structural Database. Only the molecular formula is available, also from various other sources including chemical catalogs, the PDF-2 powder diffraction database or from thousands of Web pages because this compound is a well known antibiotic.
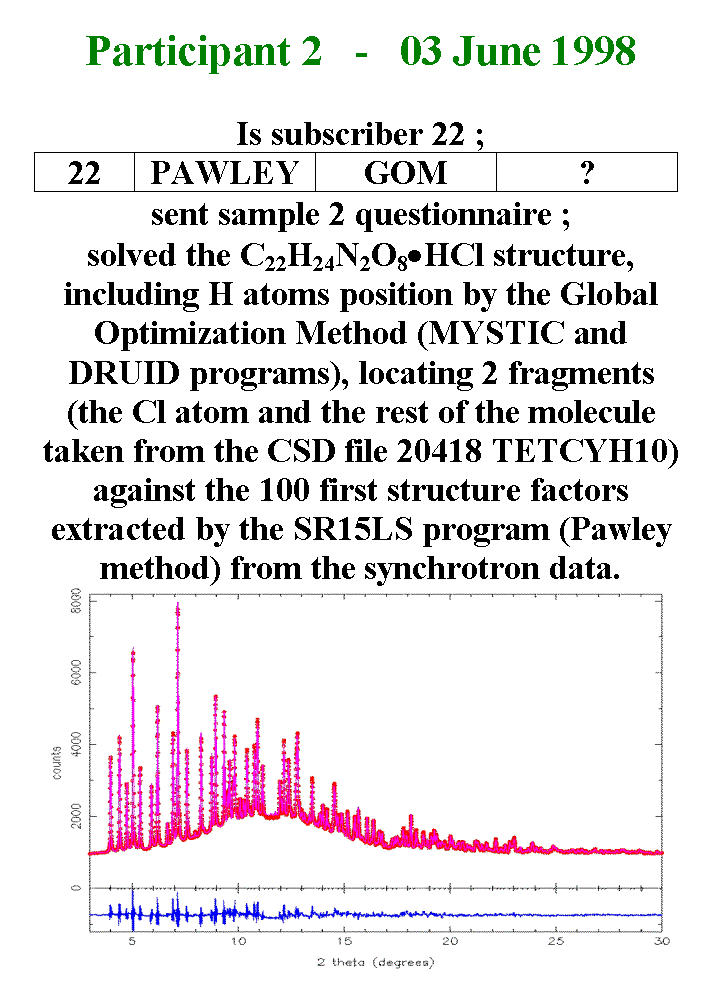
T26 - Participant 2 is the only regular subscriber having sent a successful questionnaire. Participant 2 focused his attention exclusively on sample 2 and solved its structure, including the hydrogen atom positions by the Global Optimization Method. A model for the molecule was taken from the tetracycline hydrate in the Cambridge Structural Database, removing the water. The tetracycline fragment as well as the Cl atom were positioned at random in the cell and an optimum position was searched by simulated annealing using the DRUID program against the 100 first structure factors extracted by the Pawley method from the synchrotron data. The final Rietveld refinement plot is shown here.
T27 - Participant 3 had not easy Web access and obtained the data by Email. He thought that the sample 2 would be unsolvable without the molecule connectivity and asked for it. We had anticipated that we would reply positively to such a request, so that we provided the molecular formula. Participant 3 sent filled questionnaires for samples 1 and 2, estimating finally that both of them were unsolvable. We are forced to conclude that at least 29 other regular subscribers having filled in the Web form and given their intention, also did not think that the data could be solved in 6 weeks. Anyway, they did not returned the questionnaires.
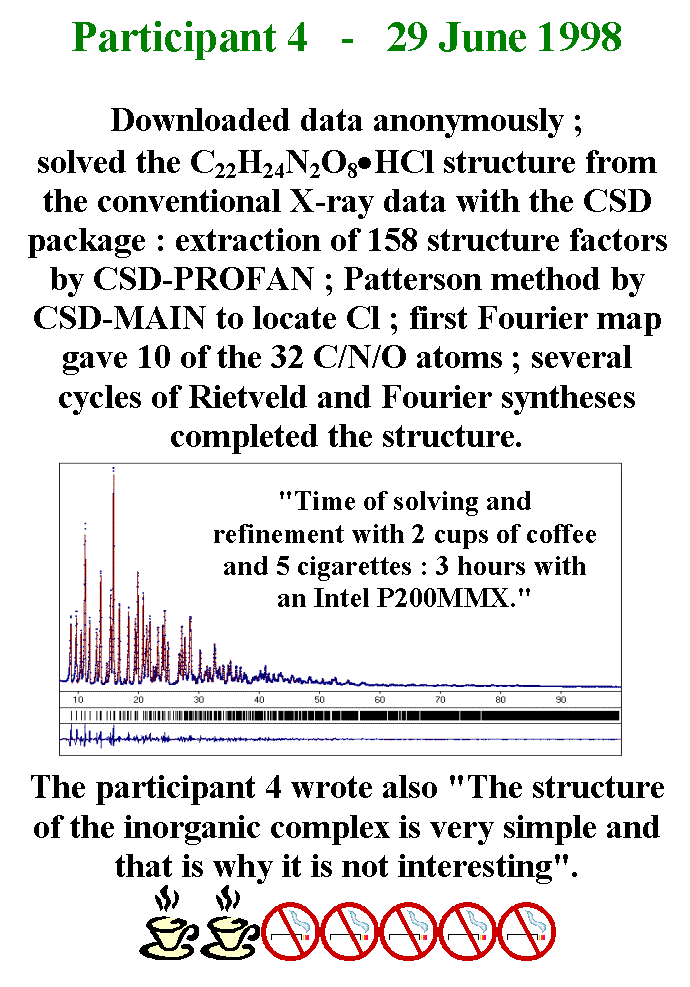
T28 - Participant 4 downloaded the data anonymously. He solved the sample 2 structure from the conventional X-ray data by using the CSD package. 158 structure factors were extracted by using the CSD-PROFAN program. The Patterson method allowed to locate the Chlorine atom by using the CSD-MAIN program. The first Fourier map produced the coordinates of ten of the other atoms. Several cycles of Rietveld and Fourier syntheses allowed to complete the structure. According to participant 4, the full time needed for solution and refinement was only 3 hours, 2 cups of coffee and 5 cigarettes with a low-end Intel PC. Participant 4 wrote also that the structure of the inorganic complex is very simple and that is why it is not interesting.
T29 - So that nobody proposed a structure for sample 1. Was it really too simple or was it too complex ? At least, it was solved by the organisers from the low-resolution powder pattern which was free of preferred orientation. The structure factors were extracted by using the FULLPROF program and the direct methods were applied to a reduced dataset by using SHELXS. A starting model was obtained in the acentric P21 space group, including the cobalt atom and ten other atoms. The structure was completed by Fourier syntheses alternated with Rietveld refinements. The job extended over 1 day, including some coffee but no cigarettes, thank you. Sample 1 is certainly not as simple as suggested by participant 4, with 15 non-hydrogen atoms in general position to be found from a powder pattern with Full Width at Half Maximum larger or equal to 0.25 two theta degrees. Here is an ORTEP view of the structure.
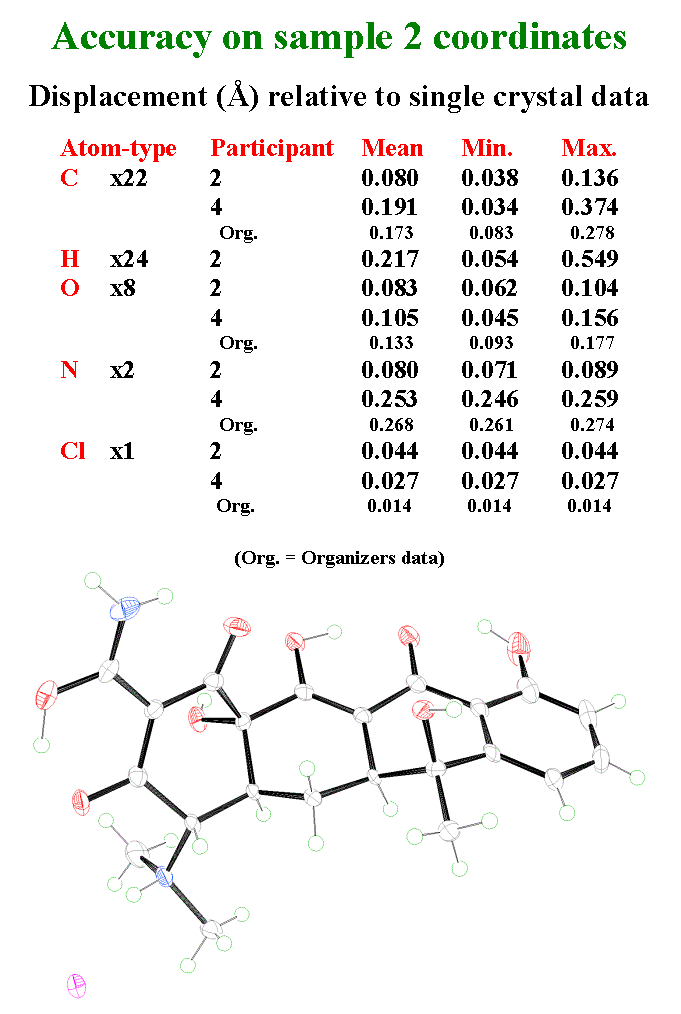
T30 - Now, some words about the accuracy of the coordinates from participant 2 and 4. A very small single crystal was taken from the sample 2 powder with size forty-per-thirty-per-twenty micrometers and data were recorded at Daresbury on the 9.8 station. The structure could be refined without any constraint, including the hydrogen atoms. Participant 2 has provided the most accurate results with mean displacements relative to the single crystal data lower by a factor 2 than those from participant 4 and from the organisers. We don't know exactly why. Even the Hydrogen atom positions are well located with a mean error of 0.2 angstroms. Here is an ORTEP view of the tetracycline hydrochloride structure. Only one hydrogen atom was not located by participant 2 : this one.
T31 - So, why ? Why 70 data downloads, 31 regular subscribers and only 4 final participants filling the questionnaires ? And why so few success ? We have not all the answers. Possibly, this is because the subscribers had not found enough time ? Or the participants believed that sample 2 was already known since it seemed to be in the Cambridge Structural Database ? Or they estimated that the sample 1 structure was too simple. But how to explain that 29 regular subscribers cancelled their intention to participate ? the only answer we have is that, really, it is not that simple to determine those structures "on demand". Anyway, your opinions are welcome.
T32 - Thanks for your attention, here is the list of people that we should aknowledge.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}