Download
McMaille version 3.0 is distributed under the GNU Public Licence
conditions.
The zipped package contains the executable for Windows 95/98/NT/XP,
as well as the FORTRAN source code (quite short and documented) and some examples
described below.
Get it : McMaille-v3.zip
The compiler used for building the executable was Compaq Visual
Fortran.
About the .hkl files :
Let the cub.hkl, hex.hkl, rho.hkl, tet.hkl, ort.hkl, mon.hkl, tri.hkl files
in the same
directory as McMaille.exe as well as your parameters .dat files. These .hkl
files contain a list of predetermined Miller indices ordered according to
a cell having a, b, c, parameters close to each other.
References
A text about McMaille is published now, to be cited in case of use :
"Monte Carlo Indexing with McMaille", A. Le Bail, Powder Diffraction 19 (2004)
249-254.
You can find a report on an indexing Round Robin in another recent paper
:
"Renewed interest in powder diffraction data indexing" J. Bergmann, A. Le
Bail, R. Shirley and V. Zlokazov, Z. Kristallogr. 219 (2004) 783-790.
Indexing Round Robin site :
http://www.cristal.org/uppw/list.html
Indexing Benchmarks :
http://www.cristal.org/uppw/benchmarks/
In case you need more, there are 2 powerpoint files available
at my Web site corresponding to congress in 2003
(ECM-21, South Africa, and SSPD03, Slovakia) :
http://www.cristal.org/ppt/LeBail-SSPD03.ppt
http://www.cristal.org/ppt/LeBail-Durban-2003.ppt
More on the method
As soon as a Monte Carlo cell proposal produces Rp < Rmaxref
~0.5 (similar definition as Rp in the Rietveld method), that cell is more
closely examined. Because a least square refinement would not be efficient,
the cell parameters are changed (NCYCLES times, see below) a bit (in the
range 0. to 0.02 Angstroms and 0. to 0.2 degrees), randomly by using the
Monte Carlo process, around their values, checking if Rp decreases. Most
of the times Rp decreases enormously, sometimes below the selected Rmax (for
keeping the cell) and Rmin (for stopping the run cause the cell could be
the right one). This cell adjustment is analogous to simulated annealing.
Moreover, a second criterium is used being that if the number of expected
peaks is explained (NDAT-NIND) with Rp > Rmaxref, that proposal cell is
examined too. This is a brute force indexing approach, very simple to develop.
Least square parameters refinements (using the old CELREF routine by Laugier
& Filhol) are performed at the end on the selected cell(s).
Some important values defined in the program are below :
Nhkl Min Nhkl Max NCYCLES NTRIED/NSOL
cubic 6xNDAT 400 200 100
rhombohedral 12xNDAT 600 500 1000
hexagonal 12xNDAT 800 500 1000
tetragonal 12xNDAT 800 500 1000
orthorhombic 20xNDAT 1000 1000 10000
monoclinic 20xNDAT 1000 2000 100000
triclinic 20xNDAT 1000 5000 100000
NDAT = Number of powder pattern peaks examined
Nhkl = Number of calculated hkl compared to the data
(read in the .hkl files)
NCYCLES = Number of random parameter small changes for a given
selected cell proposal (having Rp < Rmaxref)
NTRIED = Number of Monte Carlo events
NSOL = Number of solutions retained having Rp < Rmax
The NTRIED/NSOL ratio helps to reduce the number of retained
cells. If the value is < to the numbers listed above, then
Rmax is decreased by 5%. However, the process is not active
if NSOL < 50 and Rmax should be given negative. Avoiding being
overloaded by cell proposal is better resolved by decreasing
the control parameters W (peak width) and/or Nind (number of
non-indexed peak positions tolerated) and/or Rmaxref (the Rp
level below which a cell will be refined).
Parameters
Running McMaille (by either clicking on McMaille.exe and giving
the entry file name - no extension - or in a DOS box by typing "McMaille
name" ) requires a parameters data file. A typical data file (should be named
name.dat, name being your choice) follows :
SR2CR2O7 Title
1.54056 0.0 2 Wavelength, Zeropoint, Ngrid
1 1 1 0 0 0 Symmetry codes
0.16 6 W , Nind
3. 15. 200. 1500. 0.1 0.2 0.4 Pmin, Pmax, Vmin, Vmax, Rmin, Rmax, Rmaxref
0.2 0.2 Spar, Sang (grid search only)
20000 1 Ntests, Nruns (Monte Carlo only)
!!! A line starting by ! is ignored
11.180 345. 2-theta (or d(A)), Intensity
12.217 1120. Etc
15.835 124. 20 couples of positions and
18.709 455. intensities should cover usual
Etc cases, but more may be
necessary (max = 100)
Or, if W above is negative :
11.180 345. 0.16 2-theta, Intensity, W
12.217 1120. 0.10 Etc
15.835 124. 0.24 triplets of positions,
18.709 455. 0.16 intensities and widths
Etc
In Black box mode, the file is much shorter :
SR2CR2O7 Title
1.54056 0.0 3 Wavelength, Zeropoint, Ngrid
!!! A line starting by ! is ignored
11.180 345. 2-theta, Intensity
12.217 1120. Etc
15.835 124. 20 couples of positions and
18.709 455. intensities. You may put more
Etc but only 20 will be used.
Title : for your problem
identification.
Wavelength
: your experiment wavelength. If you used CuKalpha, you should have stripped
alpha2 before peak positions hunting.
Zeropoint
: your powder pattern zeropoint (global value including the zero due to the
diffractometer and the zero due to sample misplacement - will be added to
the data). It is recommended to have a standard compound mixed with your
sample or to apply the harmonics method for zeropoint estimation.
Ngrid : code for
the process to be applied
Ngrid
= 0 : Monte Carlo
Ngrid
= 1 : grid search
Ngrid
= 2 : both process
Ngrid
= 3 : black box mode - Monte Carlo
on all symmetries
Ngrid
= 4 : black box mode - Monte Carlo
on all symmetries + grid search
In black box mode, the next lines should be the 2-theta and intensities
couples of values, directly - see the nameb.dat files.
NOTE : grid search in triclinic is not implemented (would be too
long...)
Symmetry codes
: 6 codes allowing to select the crystal system to be explored.
1st code : if 0, no search, if 1, search in cubic
2nd code : if 0, no search, if 1, search in hexagonal/trigonal
if 2, search in rhombohedral (hex. setting)
3rd code : if 0, no search, if 1, search in tetragonal
4th code : if 0, no search, if 1, search in orthorhombic
5th code : if 0, no search, if 1, search in monoclinic
6th code : if 0, no search, if 1, search in triclinic
W : the width of the
columnar peak shape in degrees. It is recommended to choose W = 2 * FWHM,
as a minimum. Using 0.1 < W < 0.3 should produce some correct cells
for in-lab data at ~1.5 A wavelength. Using 0.05 < W < 0.15 could be
applicable to data coming from a synchrotron Facility at ~0.7 A wavelength
(extremely good peak positions are certainly required, anyway). This
parameter should reflect your data accuracy, it is close to a tolerated error.
Large values (0.30 for a copper target) give more chance to the Monte Carlo
process to find easily a minima, but the risk is to be overloaded by false
propositions. Play with it... The fact is that most of the test cases will
produce the correct solution faster with W=0.5. However, being overloaded
by cell proposal is resolved by decreasing W (peak width) or Nind or
Rmaxref.
NOTE : if W is negative, then, triplets of 2-theta, I and
Width values should be read instead of doublets of 2-theta and I values.
Moreover, these widths will be multiplied by -W (then, use W=-1 if you wish
not to change the widths, or W=-2 if you want to enlarge the widths by a
factor 2, etc).
Nind : Number of
non-indexed reflections you tolerate. Why not 2-6 for a set of 20 hkl ? Avoiding
being overloaded by cell proposal is resolved by decreasing Nind (or W or
Rmaxref).
Pmin, Pmax : minimum
and maximum cell parameters for the search. Try first 2-15 or 2-20, then,
if no solution appears, increase Pmax.
NOTE : If Pmin is negative, then it becomes possible to play more
on the individual parameter limits, and a supplementary line should be given
with 12 values :
a-min, a-max, b-min, bmax, c-min, c-max, alpha-min, alpha-max, beta-min, beta-max,
gamma-min, gamma-max.
This may allow to explore in shorter time some special cases (for instance
in monoclimic, when a and c are large and b small, the 20 first lines can
be h0l lines, so that one can fix a, c and beta and explore b on more than
20 lines).
Vmin, Vmax : minimum
and maximum cell volumes for the search. Try first small volumes 20-400, then
increase Vmax if no solution occurs.
Rmin, Rmax, Rmaxref
: Rp profile reliability factor limits.
There should be Rmin < Rmax < Rmaxref
Rmin allows to stop the search as soon as a a cell corresponding
to
Rp < Rmin is obtained - use 0.01-0.15 or up to 0.20 for
bad quality data. Choosing Rmin negative allows to avoid
any program stop before the end of the total number of
Monte Carlo events or before the total grid search end.
Rmax is the max Rp value below which a MC-refined cell is kept
in memory - use ~0.20 (or up to 0.50 if you wish). Decrease
that value if the program produces too much results (no more
than 1000 cell will be sorted, anyway). If Rmax is given
negative, Rmax will be decreased dynamically (though never
below 0.20) by the program if the NTRIED/NSOL ratio is
less than values listed above in the method paragraph.
Rmax
should not be confused with the limit Rp < 0.5 allowing to
select a cell proposal for MC-refinement. That Rp < 0.5
limit is fixed in the program, it is not applied however if a
cell proposal fits with the expected number of peak positions.
Avoiding being overloaded by cell proposal is better resolved
by decreasing the control parameters W (peak width) and
Nind (number of non-indexed peak positions tolerated), than
by decreasing Rmax manually or dynamically.
Using Rmax > 0.5 enables the Two Phase mode.
Rmaxref
will have to be close to Rmax+0.1.
Rmaxref
is the max Rp value below which a cell proposal is Mc-refined.
Use 0.4-0.5 is recommended. This is the first criterium for a cell
MC-refinement (icode = 1 in the .imp output file), the second
criterium being that if the expected number of peaks is indexed,
then the cell is MC-refined whatever Rp (icode = 2). The icode
output allows you to know how the cell was obtained.
NOTE : the line including the 2 following parameters is optional
(should not occur if NGRID = 0)
Spar : grid search
step applied to the cell parameters.
Recommended values (small values increase calculation time, but too
large values will not allow the cell to be determined) :
cubic : 0.01 or 0.005
hexagonal/rhombohedral/tetragonal : 0.01-0.05
orthorhombic : 0.03-0.20 (0.01 is best, but see the time)
monoclinic : 0.05-0.30 (0.01 is best, but see the time)
triclinic : not implemented
Sang : grid search
step applied to the cell angles.
Recommended values (small values influence calculation time) :
monoclinic : 0.05-0.20 (0.01 would be best, but see the time)
triclinic : not implemented
NOTE : the line including the 2 following parameters is optional
(should not occur if NGRID = 1)
Ntests : number
of Monte Carlo tests. Use 500-10000000000 or more.
cubic : 500-1000 should be enough
hexagonal/tetragonal : 10000-100000 should be enough
orthorhombic : 1000000 to 10000000 could be enough
monoclinic : 10000000 to 100000000 could be enough
triclinic : 1000000000 could be not enough...
NOTE : If Ntests is given negative, then the following
values will be applied,
allowing to test simultaneously several crystalline systems with
relatively coherent numbers of Monte Carlo tests :
cubic : -Ntests
hexagonal/tetragonal : -Ntests*50.
orthorhombic: -Ntests*50*50
monoclinic: -Ntests*50*50*50.
triclinic : -Ntests*50*50*50*50
This is to be used for a long overall night run. In that case, use Ntests
in the range 1000-2000, this corresponding in tetragonal/hexagonal
to 50000-100000, in orthorhombic to 2.5x106-5x106, in
monoclinic
to 125x106-250x106, in triclinic to 6.25x109-12.5x109.
Nruns : number
of Monte Carlo runs. One run will execute Ntests tests.
Due to Monte Carlo random number generation, performing 10 runs
of 1000 tests may not lead to the same result as 1 run of 10000 tests.
Anyway, Nruns = 1 could lead to the expected result.
2-theta (or d(A)), Intensity
: values obtained at the peak hunting step.
The test for identification of 2-theta or d(A) values is made by the difference
between the second and the first value. So, be careful to have a list of increasing
values for 2-theta and decreasing for d(A) values. Even if you use d values,
you are requested to choose a wavelength (because McMaille intrinsically
works on 2-theta values, it will change your d into 2-theta according to
that wavelength).
Recommended : 20 couples of values. Not less than 12.
Max : 100 couples of values.
You may play on the intensities and decrease those that seem
too high and which will represent a too large part of the total
intensity.
NOTE : If W was given negative above, then, triplets of 2-theta, Intensity
and W should be read there.
McMaille expects very accurate peak positions,
the same as the other indexing programs.
Output
McMaille produces 4 or 5 types of output files :
name.imp containing the details of the calculations
and a final sorted summary.
There are 2 verbosity levels, low and large. The large verbosity is
obtained by entering a negative wavelength (of which of course the
sign is then immediately changed).
name.ckm containing an ordered total list of the "best
cells" for the CHEKCELL
program. Note that the FoMs are not real FoMs, but are calculated
as the inverse of Rp multiplied by 5... A pseudo-FoM larger than 20
is a priori interesting, corresponding to Rp < 25%. A pseudo-FoM
close to 50 or larger may indicate the correct cell (Rp < 10%).
Depending on the cell proposals, partial lists are also built ;
name_cub.ckm : cubic
name_rho.ckm : rhombohedral
name_hex.ckm : hexagonal/trigonal
name_tet.ckm : tetragonal
name_ort.ckm : orthorhombic
name_mon.ckm : monoclinic
name_tri.ckm : triclinic
name_two.ckm : two phases mode output
name.mcm containing an ordered list of the "best cells" for CRYSFIRE.
name.prf containing the "best profile" result
(with lowest Rp), to be seen by the
WINPLOTR program. For this calculation, Gaussian peak shape is
used, having FWHM = W / 2, where W is the mean columnar width
above (given that it is recommended to use W = 2 * FWHM as a
minimum). The calculated pattern is obtained after 4 Le Bail fit
iterations (see an example).
name-new.dat produced only for NGRID=3 or 4 (black box mode), containing
control parameters for new searches with NGRID = 2 in cubic
symmetry.
The screen output delivers for each symmetry examined the first
cell proposal,
and then all the proposals which will correspond to a Rp decrease. This means
that the true cell may not appear here if a false one having a smaller Rp
value
is encountered before it. Anyway, the screen output will give you an idea
of the
smaller Rp attainable. Then look at the name.imp file and to its sorted summaries.
Strategy
McMaille is a "brute force" program that can be "almost exhaustive"
in grid search mode, provided the grid steps are very short. The only problem
is : TIME. Calculations for the triclinic case with 1000 steps for
each of the six cell parameters would lead to 1000000000000000000 tests, which
corresponds to many centuries at the current speed of 20000 steps per second
in McMaille-v3.0 (was "only" 1000 steps per second in McMaille-v2.0)... However,
an exhaustive search is quite manageable in grid search mode with a step
of 0.01 Angstrom for cubic/hexagonal/tetragonal crystal systems.
The recommendation is : First use TREOR, DICVOL, ITO, CRYSFIRE.
If no result, then apply McMaille with your fastest PC.
If McMaille is so long, and if it is suggested to apply the classical
software, what is the McMaille interest ? McMaille is rather insensitive to
IMPURITIES. Note that "impurity" means supplementary phase(s)
that do not contribute for more than 10% of the total intensity diffracted.
You should not expect from McMaille solutions for mixtures of 2 or more unknown
major phases (though...). It is obvious that known impurity peaks (identified
by a search/match process) should be removed from the list of peaks submitted
to McMaille.
Making several successive applications of McMaille is recommended.
First cubic, then hexagonal and tetragonal, or those 3 crystal systems in
one try. Then orthorhombic, if no clear solution appears at the previous
runs. Then monoclinic, if no clear solution appears at the previous runs.
Finally triclinic. The black box mode detailed below can do that for you
:
BLACK BOX MODE :
That option selected by NGRID=3 (or 4) uses a shortened input for examining
your problem in all symmetries (thus it may take one night or more...) by
using the following control parameters (in fact, these parameters are modulated
according to the estimated problem size, as guessed from the dmax values):
Symmetry max MC events Pmax Vmax
cubic V*0.5 3*dmax (3*dmax)**3 - no limit
hex/rhomb/tetra 400000 30 4000
orthorhombic 4x1000000 20 500-1000-1500-2000
monoclinic 4x10000000 20 500-1000-1500-2000
triclinic 4x1000000000 20 250-500-750-1000
Four runs in orthorhombic, monoclinic and triclinic will be made by using
different maximum volumes, successively.
Other global fixed parameters : NDAT cutted at 20 (if not less), NIND = 2,
Pmin = 2., Vmin = 8., W = 0.30*wavelenght/1.54056,
SPAR = 0.02, SANG = 0.05, Rmin = 0.02, Rmax = 0.15, Rmaxref = 0.40
Dmax is the d value for the first peak position at low diffraction angle.
This black box mode could solve simple cases. If not, using the manual modes
(NGRID = 0, 1, or 2) would be necessary, enlarging the above cell parameters
and volume limits. Trying first in cubic symmetry (this is why the name-new.dat
file is made for the cubic case), and then going to lowest symmetries if
no result.
For recognizing the very
best solution in a black box mode output, you have to find, in principle,
the cell proposal corresponding to the smallest Rp with highest symmetry
and smallest volume, indexing the largest number of peaks. Not always an
easy task... so, open your eyes ! Then check your choice(s) by the Chekcell
program and by whole pattern fitting by the Pawley or Le Bail methods (Fullprof,
Gsas, Rietica, Maud, etc, etc).
FASTER PRELIMINARY TEST :
You may well make the first tries by using a small data set of only NDAT
= 12 peak positions, and a large W value (0.5 at 1.54A, or 0.25 at 0.7A),
together with Rmax =0.5 and Rmin = 0.01, and a number of non-indexed peaks
of 2 or 1. You may well obtain the correct indexing in that way, very fast
(speed will be increased by a factor 2 or 3 due to the Nhkl decrease - see
above the Nhkl definition) . If no result, go
to at least NDAT = 20, and use conditions as recommended in the parameters paragraph above.
Repeat several Monte Carlo runs if nothing is produced (several
Monte Carlo runs will not use the same random number sequences, and will
not examine the same combinations of cell parameters). This is essentially
a question of chance...
Because calculations can be extremely long if you use the grid search
procedure with small steps, a WARNING occurs at the beginning of McMaille
runs giving an estimation of the calculation time (on the basis of 20000 tests
per second obtained with an Intel Pentium IV 2.4GHz processor). Note that
these 20000 tests per second were estimated for a cubic case with Nhkl =
400 in the cub.hkl file. In the more complex triclinic case, the test number
with Nhkl = 1000 in tri.hkl decresases to 7000 per second. You may obtain
more tests per second by decreasing Nhkl (~40000 per second in cubic with
Nhkl = 100). The speed will also strongly depend on the number of peak positions
selected (20 recommended). In order to have a real idea of the needed time,
make a small run (100000 Monte Carlo events, for instance) and extrapolate
to the large run.
LARGE CELLS
If you want to index proteins, then :
- if you give 2-theta values as peak positions, do not change them but divide
the wavelength by 10, so that the cell parameters proposed by McMaille will
be divided by 10 as well.
- if you provide d(A) values as peak positions, divide them by 10 and choose
a wavelength very short (0.06-0.15 A). So, the cell parameters provided by
McMaille will have to be multiplied by 10 in order to recover the true values.
TWO PHASES MODE (use cautiously !):
In desperate cases, this mode will propose to interpret the data with two
phases. This mode is enabled if Rmax > 0.5. This is quite logical since
you will expect that each single phase will represent less than 50% of the
total intensity of the powder pattern. Recommended values for Rmax and Rmaxref
are 0.6 and 0.7, respectively. You will have to supply at least 30 peak positions,
and the number of tolerated non-indexed peaks will have to be high (say 18
non-indexed for 30 peaks). In this mode, a quite large number of cells will
be tested so that the speed is considerably decreased. Waiting for faster
computers, it is suggested to limit that mode to cubic/hexagonal/rhombohedral/tetragonal/orthorhombic.
More than 1000 cells will easily appear and force the run to stop. A list
of couples of cells that may explain together a maximum of peak positions
is provided at the end of the .imp file. Two examples are distributed with
the test files (mixture1 and mixture2). That mode may work or not, of course...
NOTE : pressing the K keystroke (capital letter - for Kill) will
stop the program a few seconds later, saving the current results.
Examples
The test samples attached with the McMaille package (testn.dat)
come mainly from the TREOR and DICVOL distribution package tests (using arbitrarily
intensities set to 100.), plus some other example like Y2O3,
NAC, and the samples 1-3 from the SDPDRR-2 Round Robin. Running
them on your own PC should produce the solutions. Examples of time (Pentium
IV 2.4GHz) needed by McMaille for its test files are below (all tests
by Monte Carlo, not grid search) :
Cimetidine (cim.dat) : monoclinic - 9 seconds
Rp Vol
Ind a
b
c alpha
beta gamma
0.026 1279.113 21 10.3893 18.8215
6.8215 90.000 106.477 90.000
M(20) = 503.4126
F(20) = 1333.414
( 4.9996987E-04,
30)
NAC (nac.dat) : cubic - < 1 second
Rp Vol
Ind a
b
c alpha
beta gamma
0.046 1078.129 20 10.2539 10.2539
10.2539 90.000 90.000 90.000
M(20) = 93.76609
F(20) = 66.04718
( 5.9375260E-03,
51)
SDPDRR2 Sample
1 (sample1.dat) : monoclinic - 23 seconds
Rp Vol
Ind a
b
c alpha
beta gamma
0.045 651.662 20
8.5301 7.4004 10.3260 90.000 91.336 90.000
M(20) = 46.97108
F(20) = 75.49640
( 5.4063937E-03,
49)
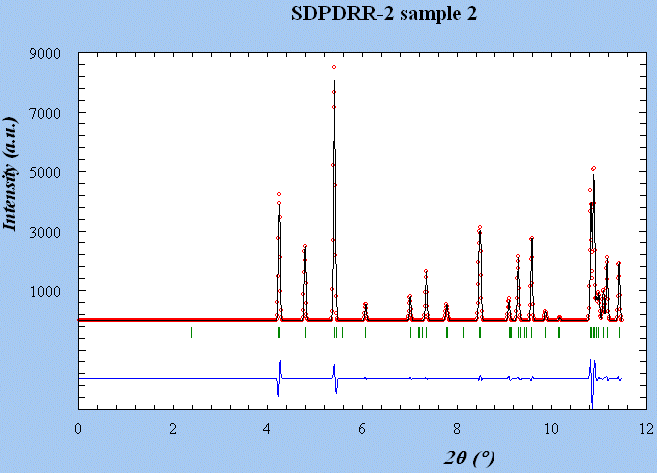
SDPDRR2 Sample
2 (sample2.dat) : monoclinic - > 6 minutes
Start : 17-Oct-2002 18 hour 35 min 36 Sec
Rp Vol
Ind a
b
c alpha
beta gamma
0.033 1760.121 22 19.9496
8.1937 11.2441 90.000 106.736 90.000
M(20) = 101.8119
F(20) = 588.4827
( 9.1853255E-04,
37)
SDPDRR2 Sample
3 (sample3.dat) : cubic - 1 second
Rp Vol
Ind a
b
c alpha
beta gamma
0.056 6735.840 24 18.8856 18.8856
18.8856 90.000 90.000 90.000
M(20) = 149.7873
F(20) = 512.6646
( 7.2244194E-04,
54)
Test 1 - Cd3(OH)5(NO3) (test1.dat) - orthorhombic - 3 seconds
Rp Vol
Ind a
b
c alpha
beta gamma
0.037 378.227 20 11.0279
3.4202 10.0277 90.000 90.000 90.000
M(20) = 126.0809
F(20) = 183.3493
( 3.7614279E-03,
29)
Test2 (test2.dat) - tetragonal - < 1 second
Rp Vol
Ind a
b
c alpha
beta gamma
0.083 1186.855 25 11.1886 11.1886
9.4809 90.000 90.000 90.000
M(20) = 32.65442
F(20) = 58.72479
( 9.4603244E-03,
36)
Test3 (test3.dat) - orthorhombic - 5 seconds
Rp Vol
Ind a
b
c alpha
beta gamma
0.101 1154.716 25 11.3318 9.2362
11.0328 90.000 90.000 90.000
M(20) = 17.36584
F(20) = 29.50007
( 1.0593196E-02,
64)
Test 4 : monoclinic - less than 1 minute
Rp Vol
Ind a
b
c alpha
beta gamma
0.077 684.950 25 6.2461
12.4695 9.1917 90.000 106.911 90.000
M(20) = 52.42331
F(20) = 110.0724
( 6.4892345E-03,
28)
Test 5: (NH4)2S2O3 - monoclinic - 16 seconds
Rp Vol
Ind a
b
c alpha
beta gamma
0.094 582.592 25 8.8043
6.4951 10.2231 90.000 94.757 90.000
M(20) = 33.04150
F(20) = 59.24461
( 7.1826265E-03,
47)
Test 6 : triclinic - small cell - < 2 minutes
Rp Vol
Ind a
b
c alpha
beta gamma
0.079 182.342 25 7.6256
5.5093 5.1169 89.828 74.979 62.441
M(20) = 37.16255
F(20) = 53.50393
( 1.2460144E-02,
30)
Test7 - cubic ??? - < 1 second
Rp Vol
Ind a
b
c alpha
beta gamma
0.110 13743.956 23 23.9536 23.9536 23.9536
90.000 90.000 90.000
M(20) = 6.623881
F(20) = 14.28700
( 1.7071631E-02,
82)
Test 8 - monoclinic - < 2 minutes
Rp Vol
Ind a
b
c alpha
beta gamma
0.098 149.517 20 5.0750
5.8569 5.0319 90.000 91.444 90.000
M(20) = 50.94925
F(20) = 54.74235
( 1.0148551E-02,
36)
Test 9 - triclinic - < 1 minute
Rp Vol
Ind a
b
c alpha
beta gamma
0.069 984.080 20 7.0828
18.8631 8.7848 117.123 94.043 71.092
M(20) = 52.09227
F(20) = 135.3708
( 5.2765133E-03,
28)
Y2O3 - cubic - < 1 second
Rp Vol
Ind a
b
c alpha
beta gamma
0.073 1190.426 19 10.5983 10.5983
10.5983 90.000 90.000 90.000
M(20) = 136.5140
F(20) = 96.67932
( 3.9031976E-03,
53)
See also the nameb.* files which are corresponding to the Black Box mode.
See also the mixture1 and mixture2 files corresponding to the Two Phases mode.
In mixture1.imp (2 cubic phases), the correct couple of solutions
appears in 15th position :
Rp2 Vol Ind Nsol a b c alpha beta gamma
0.113 1078.288 29 7 10.2544 10.2544 10.2544 90.000 90.000 90.000
0.165 1190.411 14 7 10.5982 10.5982 10.5982 90.000 90.000 90.000
In mixture2.imp, (one tetragonal + one orthorhombic phase), the correct solution
is the 1st :
Rp2 Vol Ind Nsol a b c alpha beta gamma
0.259 1188.120 30 13 11.1880 11.1880 9.4919 90.000 90.000 90.000
0.106 378.244 15 4 10.0276 3.4206 11.0274 90.000 90.000 90.000
Times may be different on your machine (could be less or more, this is Monte
Carlo... you need chance).
In 15-20 years, computers will be 210 to 213
faster (x1000 to x8000 faster), at least, probably. Even grid search in triclinic
will be manageable.
To do
I have done a lot already, wasting randomly considerable time ;-)...
But improving the cell proposal lists by more detection of redundant cells
(supercells, etc) has to be done, providing a more clean list of possible
cells, including the Bravais lattice recognition.
Send your comments, ideas and bug reports
(thanks to L.M.D. Cranswick for many of them)
to :
Armel Le Bail
- September/October 2002
|
{kind=link}