SDPD Internet Course
Semaine 6
Solution de structure par méthode Patterson, directe ou par localisation de molécule - niveau 1 : méthodes conventionelles
Lectures
Lisez avec attention le chapitre 3.3 du didacticiel SDPD et/ou lisez le texte (anglais) de la conférence Kunming Workshop, même chapitre (milieu de la 3ème partie). Plusieurs exemples de détermination de structure par approche classique sont donnés (cependant, la version anglaise du didacticiel SDPD est plus complète). Vous devriez essayer de reproduire les résultats obtenus pour ces exemples (Na2C2O4, [Pd(NH3)4]Cr2O7, t-AlF3, beta-BaAlF5 et cimetidine C10H16N6S) avant de tenter de résoudre l'exercice proposé en bas de cette page.
Regardez de près les "SDPD Round Robin" 1 (1998) et 2 (2002). Une série de problèmes est disponible avec diverses solutions proposées par des compétiteurs sur un plan international.
Lisez les chapitres de livres :
B4 : Chapitre 2, pages 61-137 et
Chapitre 5, pages 319-397.
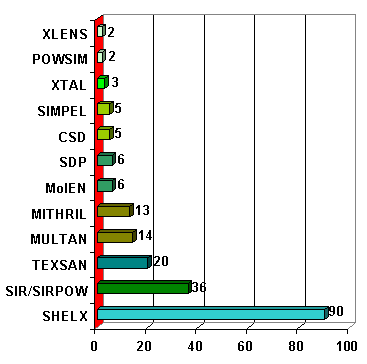
Cette session est limitée à l'utilisation des méthodes conventionnelles, c'est-à-dire par application de logiciels dédiés aux études structurales des monocristaux : principalement SHELXS pour les méthodes directes et de Patterson. Une liste (presque) complète de logiciels qui ont été utilisés pour déterminer des structures par la méthode des poudres est proposée dans la banque de données SDPD (Il n'y a cependant pas de lien hypertexte vers chacun de ces logiciels). Les statistiques de 1998 placent SHELXS largement en tête :
Logiciels à télécharger (éventuellement)
Il vous avait été recommandé en 2ème semaine de vous procurer, auprès de leurs auteurs, les dernières versions de SHELX, WinGX et SIR97. Vous pouvez souhaiter contrôler SHELX par l'intermédiaire de WinGX ou d'un autre logiciel. Vous pouvez aussi éventuellement vous intéresser à d'autres logiciels comme, par exemple :
Caos par S. Cerrini & R. Spagna
Crunch par R. de Gelder et R.A.G. de Graaff (méthodes directes), seulement sur machine Unix (et Linux). Voyez un didacticiel au CCP14.
Crystals par D. Watkin (pages du CCP14).
Dirdif par P. T. Beurskens, Patterson et méthodes directes appliqués à des facteurs de structure différence. Une version pour Windows est disponible : WinDirdif (pages du CCP14).
MITHRIL par C. Gilmore, inclus dans la suite logicielle commerciale TeXsan.
Jana par V. Petricek et M. Dusek, couvre toutes les tâches usuelles de l'analyse structurale, à l'exception du graphisme et des méthodes directes (pages du CCP14).
Multan par P. Main, Méthodes directes, inclus dans de nombreux logiciels commerciaux (?). Vous pouvez télécharger le code source Fortran d'une vieille version (1980) avec le manuel correspondant.
SDP commercial
SnB Shake and Bake
TeXsan commercial par MSC (Rigaku)
UNICS par Sakurai & Kobayashi
Xlens par J. Rius
etc.
Notez que GSAS réalise des analyses de Patterson.
Autres logiciels
Un petit programme peut être parfois très utile pour éliminer ces "|Fobs|" correspondant à des réflexions qui chevauchent de trop près d'autres réflexions.
Overlap par A. Le Bail
Certaines suites logicielles, appliquant la méthode de Patterson ou les méthodes directes, incorporent des traitements spéciaux des données de diffraction de poudre (Expo, Dorees dans Powsim, FIPS, Focus....), mais ce sera le sujet de la semaine prochaine de cours.
Logiciels de visualisation de structure
La visualisation des modèles de structure complets ou partiels est une des clés du succès. Vous devez donc vous procurer cette semaine quelques logiciels parmi ceux de la liste suivante (logiciels commerciaux exclus, mais il y en a d'excellents évidemment, comme Diamond par exemple) :
Ortep-3 pour Windows par Louis Farrugia (ortep32.zip - il faut demander une licence à l'auteur), basé sur Ortep-3 par Michael N. Burnett et Carroll K. Johnson.
Struplo pour Windows par Louis Farrugia (struplo.zip - il faut demander une licence à l'auteur), basé sur Struvir par Armel Le Bail (lui-même basé sur Struplo par R.X. Fischer).
Struvir par Armel Le Bail, version DOS, code source Fortran disponible, basé sur Struplo by R. X. Fischer.
Platon pour Windows
par Louis Farrugia (pwt.zip), basé
sur Platon par Ton
Spek.
Schakal a été récemment mis en accès libre aux chercheurs académiques (CD: Schakal.zip).
Chime basé sur Rasmol, plug-in essentiel pour votre butineur (ch203w32.exe).
DrawXTL par Larry Finger et Martin Kroeker (drawv31.exe).
Jamm par John N. Huffman.
JSV par Steffen Weber.
Ortex par Patrick McArdle.
PowderCell par Werner Kraus et Gert Nolze (pcw23.exe).
Rasmol par R. Sayle et Protein Explorer.
WebLab viewer lite par Accelrys (WLViewerLite40.exe).
Xtal-3D par M. hewat (VRML file builder).
Xtaldraw par K.L. Bartelmehs.
Pour plus de détails, voyez la page Web du CCP14 sur la question. Les logiciels commerciaux y sont nombreux également (Atoms, Crystal Maker, Diamond, Schakal, etc).
Le language VRML (Virtual Reality Markup Language) est un moyen pratique d'accéder à une visualisation de vos modèles structuraux en 3D (fichiers .wrl produits par STRUVIR/WinStruplo ou par Diamond par exemple). Une liste d'outils VRML pour la cristallographie est disponible. Votre butineur doit être équipé d'un visualiseur VRML tel que CosmoPlayer.
Suggérez nous vos programmes
favoris si ils ne sont pas cités ici - merci.
Exercice
Les semaines précédentes, vous avez extrait les "|Fobs|" de deux composés inconnus synthétisés dans les systèmes NaF/CrF3 et PbF2/CrF3/HFaq.
Essayez maintenant d'obtenir un modèle structural pour ces phases, un modèle initial pas forcément complet mais suffisamment important pour vous permettre de démarrer un affinement par la méthode de Rietveld. Mais, à ce stade, n'utilisez pas la méthode de Rietveld, contentez vous d'obtenir le modèle initial. Essayez d'obtenir le modèle le plus large possible à partir des "|Fobs|" (jeu de données complet ou partiel) en construisant et interprètant une Patterson et/ou en appliquant les méthodes directes et le programme d'affinement de structure sur monocristal de votre choix. Vous pouvez calculer une Fourier différence, pour essayer de localiser un maximum d'atomes. Vous pouvez soit utiliser vos propres "|Fobs|" ou bien ceux qui vous sont fournis ici en format pour SHELXS format (carte HKLF 3) : na5.hkl et pbcr.hkl (le fichier .fou de Fullprof renommé, texte enlevé, auquel on ajoute une ligne de zéro à la fin). Vous pouvez aussi avoir besoin des fichiers résumé .sum de Fullprof qui correspondent à ces extractions de facteurs de structure : na5.sum et pbcr.sum contenant les paramètres de maille et le groupe d'espace, au cas où vous souhaiteriez utiliser le programme OVERLAP. L'achèvement de la détermination de structure par alternance d'affinements Rietveld/nouvelle extraction de "|Fobs|"/synthèse de Fourier, sera le sujet des sessions 9 et 10 : donc, ne le faites pas maintenant. L'objectif de cette session est de voir jusqu'ou vous pouvez aller avec cette première extraction des "|Fobs|", parce que c'est toujours la meilleure chose à faire plutôt que d'essayer trop tôt de tester par la méthode de Rietveld un modèle de structure insuffisant. N'utilisez pas non plus le logiciel EXPO cette semaine (il sera vu la semaine suivante), car nous voulons que vous "mettiez les mains dans le cambouis" (bien que la plupart des logiciels de Patterson et méthodes directes fonctionnent en mode "boite noire") avant d'essayer les logiciels encore plus "automatiques".
Qui sait, vous pourriez déterminer déjà la structure complète de ces deux phases à partir de ces "|Fobs|" ? Les résultats demandés sont les coordonnées atomiques de tous les atomes constituant votre meilleur modèle initial. Donnez aussi les paramètres de maille et le groupe d'espace (les choix d'orientation des mailles peuvent être différents). Expliquez en pas plus de 20 lignes comment vous avez réalisé le travail.
Une connaissance de la cristallochimie
spécifique d'une phase est absolument nécessaire avant de se
lancer dans sa détermination de structure (cependant, gare aux idées
préconçues). Cr3+ dans les fluorures est rarement
dans une autre coordinence que 6 (octahèdre), avec des distances interatomiques
Cr-F de l'ordre de 1.9 Angstrom. Na+ peut adopter des coordinences
6-9 dans les fluorures avec des distances Na-F ~2.2-2.9 Angstroms. Pb2+
adopte des coordinences 7-10, avec Pb-F ~ 2.4-3.0 Angstroms. Les distances
standard F-F sont voisines de 2.5-2.9 Angstroms.
Logiciels sélectionnés pour réaliser la correction officielle
Fullprof pour extraire les "|Fobs|";
Overlap pour construire des jeux de données réduites; SHEXLS-97
pour les méthodes Directes et de Patterson; Ortep-3 pour Windows et
Struplo/Struvir pour les dessins et la visualisation; Cosmo Player + Netscape
pour visualiser les structures 3D en VRML; SHELXL-97 pour l'affinement de
structure.
La semaine suivante sera consacrée
à la solution de structure,
niveau 2 (approches spéciales).
Bonne chance !