SDPD Internet Course
Week 10
Structure completion and refinement by the Rietveld method, part 2.
Lectures
When and how to stop a Rietveld refinement is a quite difficult question to which you will be able to anwer by yourself after years of experience. Should you refine the thermal parameters anisotropically sometimes, at least for some heavy atoms ? Should you try to find hydrogen atoms and should you include them in the refinements ? When are restraints and constraints applicable ? What is a good ratio of parameters/reflections numbers with the Rietveld method ? Are the extracted size and microstrain parameters, possibly with anisotropic effects, dubious or not ? Etc. Acceptance or rejection of a manuscript will depend on your choices. You should not go too far from reasonable limits, which are certainly difficult to define exactly...
Preferred orientation and anisotropic line broadening are the two main problems (if the sample is pure, the resolution is the best as possible, and the statistics are fine) which could limit the quality of your refinements, and also could disallow you to determine a structure from powder diffraction data.
Extracting physical parameters, characterizing the size and microstrain effects, from a powder pattern by using the Rietveld method is a risky task. Two reference books in that topic were recently published :
B6- Defects and Microstructure Analysis
by Diffraction,
R.L. Snyder, J. Fiala and H.J. Bunge,
IUCr Monographs on Crystallography, Vol
10,
Oxford Science Publications, 1999.
B9 - Fundamentals of Powder Diffraction
and Structural Characterization of Materials
Vitalij K. Pecharsky, Peter Y. Zavalij
Kluwer Academic Pub., 2003.
Several chapters in that book concern the Rietveld method. However, it contains mainly the proceedings of a 1995 congress in Slovakia "Size-Strain'95". You will have an idea about the most recent views on microstructure analysis through the Rietveld method by reading a recent conference (CD) given at the Sixth International School and Workshop of Crystallography, 22-27 January 2000, Ismailia, Egypt. The Size/Strain Round Robin (see the full article, see also a detailed contribution to that round robin (CD)) is worth to be examined (though the experimental example presented is too simple : cubic, isotropic line broadening, size effect only). Have also a look at a text (CD: PDF) in which the Rietveld method is applied to an orthorhombic phase, with anisotropic size and strain effects. Read how microstructural effects are undertaken in Fullprof (CD: PDF).
Software to download
You may have to deal with peculiar problems that are precluding you to present complete results. For instance, not locating hydrogen atoms may have an effect on your fit by more than 5% on Rp, Rwp and RB reliability factors when dealing with organic compounds. But you may have difficulties to locate these hydrogen atoms by Fourier difference syntheses, even from very good synchrotron data (there would not be any problem to locate D atoms by neutron powder diffraction - except that you have to deuteride the sample, and if the structure is not too complex). A possibility is to guess the hydrogen atom positions and to add them for refinements with constraints/restraints. Some programs can propose H atom positions :
Babel, useful for data format exchange, and locating H atoms, with a Windows front-end (CD : babelwin.zip);
Xhydex for locating H atoms in hydrides ;
Mol2Mol can do hydrogen location (commercial) ;
Molden seems to be able too ;
WebLab viewer from Accelrys/MSI (CD : WLViewerLite40.exe);
VMD ;
Gromacs ;
Molden doesn't handle the effect of different pH's ;
SHELXL, already introduced ;
Etc.
Such programs are not 100% efficients as will be shown below for the BABEL application to the tetracycline hydrochloride compound (SDPDRR) (CD):
Hydrogen atom positions,
as located from the single crystal data
:
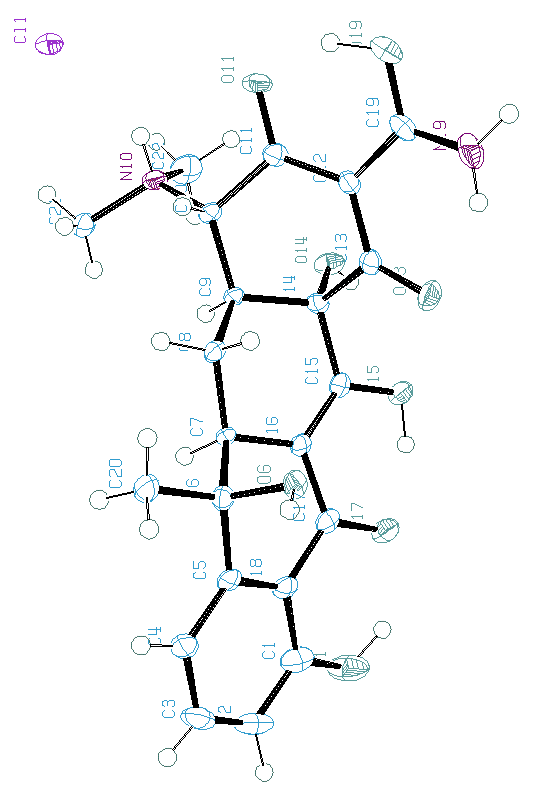
as proposed by BABEL :
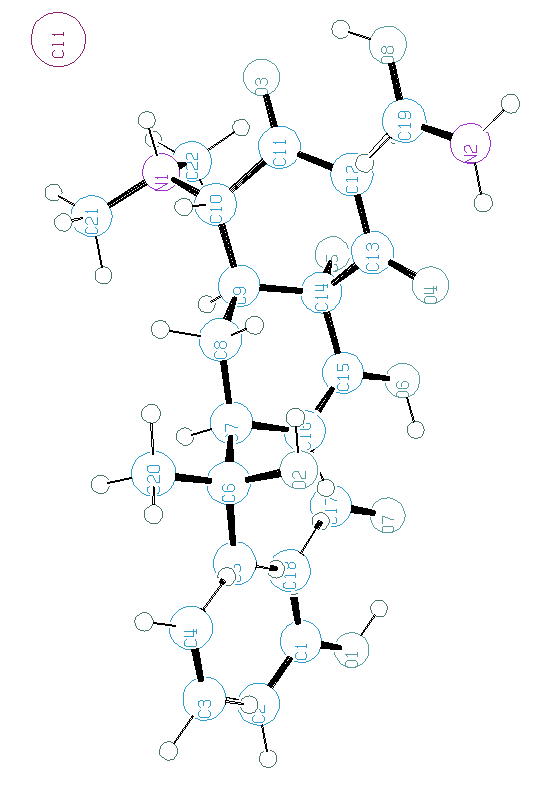
Optimization of a molecular model before going to refine with constraint/restraints or as rigid body could be quite useful. This is a bit out of the scope of this SDPD course, but think to it. Programs well known by chemists can do that (SPARTAN, GAUSS...).
About microstructures, many Rietveld programs allow some approach. This is a domain where you should take your own responsibilities because we are there at the border of recognized knowledge. Among programs which may be useful, one may cite GSAS, FULLPROF, already listed last week, and WinMProf, among others (which has some of the most recent options allowing to treat anisotropic line broadening) (CD : InstallWMP.exe).
Exercises
1- Complete the structure determination and refinement of the pyrene derivative (see week-8). Try to locate the hydrogen atoms. Then produce the best refinement you can. Decide if you should use constraints or not.
2- Try to do your best by the Rietveld method applied to the synchrotron powder pattern of Norbornane (norbornane.dat, or norbornane.pds, ready for WinMProf) C17H12 at 50K (A.N. Fitch and H. Jobic, J. Chem. Soc. Chem. Commun. (1993) 1516-1517), starting from the CSD entries (norbor.txt, norbor.ps). Wavelenght 1.39839 A. What are the main problems with this powder pattern ?
In each case, explain briefly your strategy and produce the .sum file if you used FULLPROF, or any equivalent summary of the refinement from another Rietveld program (including all refined parameters, their estimated standard deviation and the reliability factors).
Main programs selected for the correction
BABEL, FULLPROF, SHELXL, ORTEP,
WinMProf (for exercise 2)...
Two weeks will be reserved now for
the final examination.
We may start those two weeks at any date
of your choice, but we ask for receiving your results in due time.
Ask us for your total number of points
already cumulated during the course, if we forget to give it.
Good luck !